Version 2023 of ChemAnalytical Workbook adds new features, including expansion of data that can be exported to JSON format and expanded database search options among other improvements. We have also improved existing functions. Read below for details, and contact us for help upgrading your software.
Enhanced Record Set Management
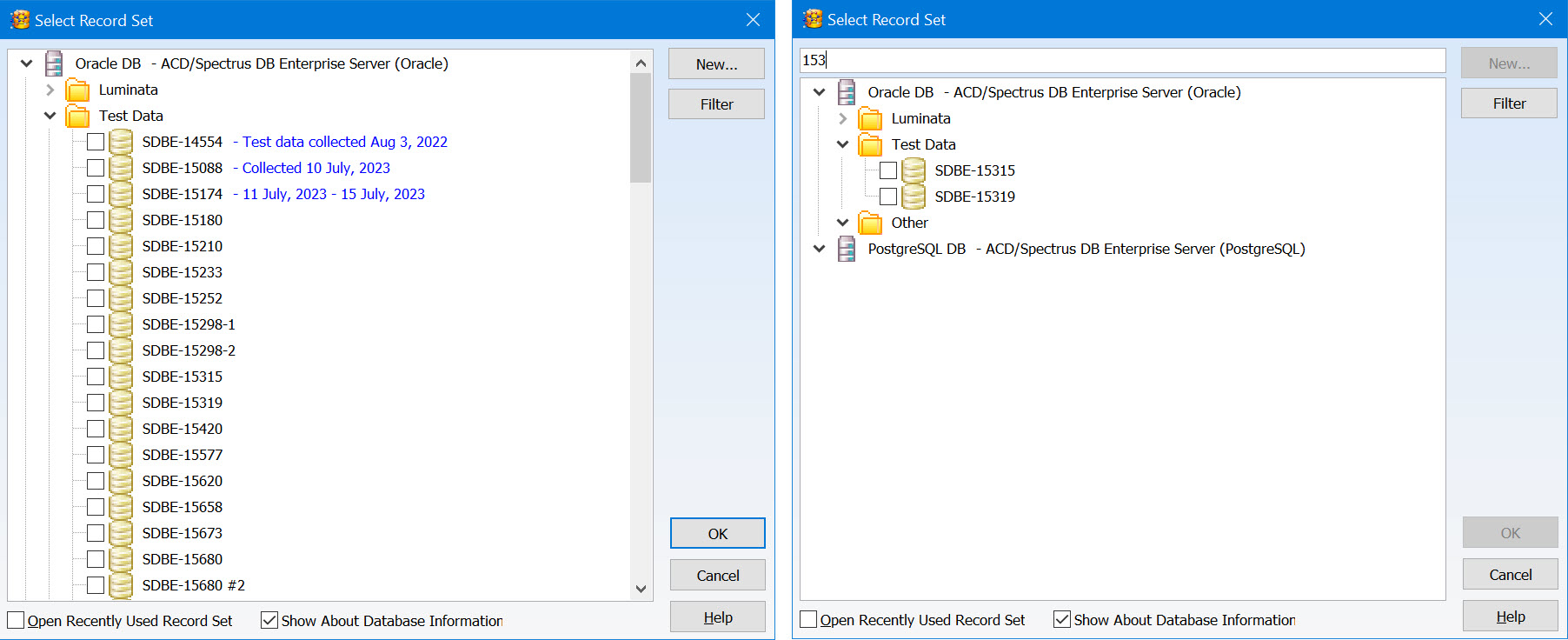
You now have additional features to help organize and review record sets, making it easier to find what you are looking for.
- Organize record sets into a tree layout using the Enterprise Server Management Console for greater control
- Add descriptive information to be displayed when you select a record set
- Text search is now available when reviewing record sets
Permission Management
Record owners can no longer open record sets by default. These permissions can be edited by a system administrator.
Expanded Database Search Options
You now have more tools for finding the data you need within database records.
- Search for records where a given table exists or not
- Common mass spectrometry related queries can now be done for a remote database
- Column names are now searchable for all tables in database records
- Perform further searches within the results of a previous search
Visualize Your Graph Data More Easily
- Customize the size of data points
- Highlight records from a given filter
- Review large data sets more easily through faster display speed
Improved Peak Naming for xC/UV/MS Data
Fine-tune peak naming for xC/UV/MS data with new options to:
- Automatically name your peak as a prefix + (optional) number suffix or use selected attributes (i.e., metadata, spectral parameters, etc.)
- Import peak names from your CDS
- Transfer peak names to the Table of Components and keep user-defined names
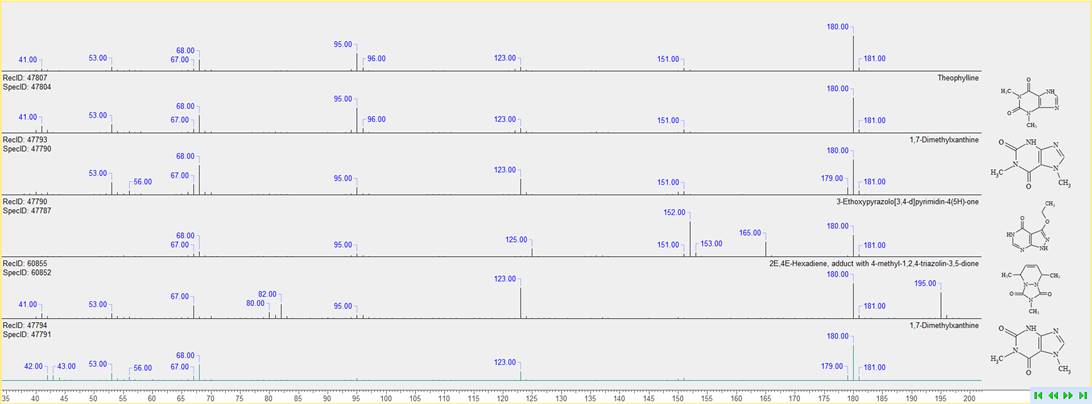
Consolidated Display of Results from Mass Spectral Search
You can now review mass spectral search results more conveniently and find the correct match through concurrent display of the query spectrum with hit spectra, structures, and metadata.
Include Integral Assessment in Structure Verification
You can now assess how well the experimental 1H integral values match those predicted for the complete structure using the integral maximum and standard deviations in the Structure User Data table.
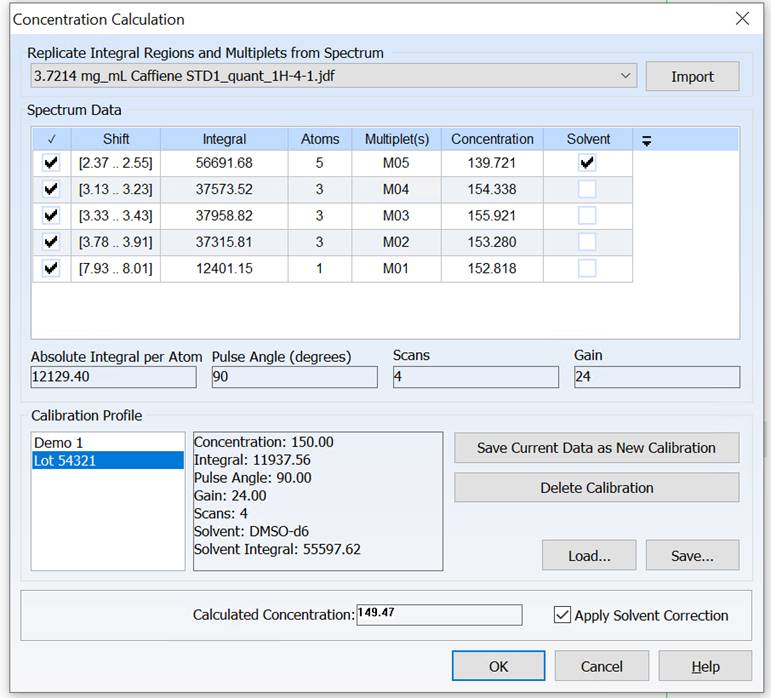
Adjust Solvent Integral to Minimize Error in External Standard qNMR
You can now compensate for error introduced by variability between NMR tubes when using an external standard for qNMR by normalizing the solvent integral in your spectrum with respect to that of the calibration spectrum.
- We improved data import from:
- Agilent OpenLab ChemStation and Bruker LC/MS ToF DAD traces—DAD data and extracted wavelengths can now be imported and viewed
- We now support Shimadzu LabSolutions CDS data from LCMS-9050 Q-TOFs
- We now support import from archives (i.e., *.zip)
- Thermo Scientific Chromeleon CDS add-on is improved to display m/z values for SIM data
- OpenLab CDS add-on now uses a single processing method to export all datasets in the OpenLab CDS repository
- We have expanded the data you can export to JSON format for use in data science and modeling
- Export hyphenated xC/UV/MS data
- Export processed 1D and 2D NMR spectra, including peaks, multiplets, and assignments
Improved Peak Detection and Integration for Hyphenated Data
You now have new options for the detection and integration of peaks:
- Specify an m/z range for spectral searching allowing the exclusion of extraneous ions and improving HQI (hit quality index) score
- Set a minimum area (% of max) threshold
- Combine adjacent and overlapping peaks by maximum resolution, and maximum number of peaks, and apply them to a specified region
- Apply minimum and maximum FWHM thresholds to flat chromatogram traces
- Equalize liftoff and touchdown heights
Account for Blank Injections for Sample Cleanup in LC/MS Data
You can now subtract flat chromatogram curves from one another to account for blank injections.
Ease of Use Improvements
- Add known fragment structures to unassigned MS peaks when atom connectivity differs from the parent structure
- Select and review the component formulae before assigning MS data
- Set method details (i.e., gradient, flow rate, etc.) once and apply across all chromatographic traces