August 1, 2018
by Mikhail Elyashberg, Leading Researcher, ACD/Labs
Oroidin
Oroidin (C11H10Br2N4O) is a highly proton-deficient bromopyrrole (1) isolated from the sponge Agelas oroides.
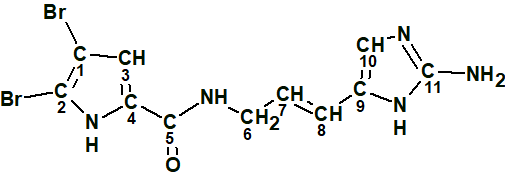
1
The ratio of the number of skeletal atoms, nsk=18, to the number of hydrogens, n(H)=10, is equal to almost 2, which is a characteristic feature of challenging structure elucidation problems. This compound has been extensively studied by IR, NMR, X-ray crystallography and was confirmed by direct synthesis [1-3]. A CASE solution to this problem was chosen for testing the capability of DFT based NMR chemical shift prediction to distinguish the correct structure when empirical prediction methods fail to suggest the structure reliably. The experimental 1D and 2D NMR data obtained for structure 1 in [1] were entered into ACD/Structure Elucidator (Table 1).
Table 1. Spectroscopic 1D and 2D NMR data for Oroidin (1).
| C/X Label | δC | δC calc* | XHn | δH | M, J | COSY | H to C HMBC |
| C 1 | 98 | 97.8 | C | ||||
| C 2 | 105 | 104.2 | C | ||||
| C 3 | 113 | 113 | CH | 6.88 | s | C 2, C 4 | |
| C 4 | 128 | 124.7 | C | ||||
| C 5 | 158.8 | 159.58 | C | ||||
| C 6 | 39.8 | 40.43 | CH2 | 3.94 | dd,5.5,4.9 | 6.13, 12.79 | C 8, C 7, C 5 |
| C 7 | 127 | 126.6 | CH | 6.13 | dt,16.4,4.9 | 3.94, 6.20 | C 6, C 8, C 9 |
| C 8 | 116 | 114.26 | CH | 6.2 | d,16.4 | 6.13 | C 6, C 9, C 7 |
| C 9 | 125 | 128 | C | ||||
| C 10 | 111 | 110.8 | CH | 6.98 | s | C 8, C 9, C 11 | |
| C 11 | 148 | 147.5 | C | ||||
| N 1 | NH | 11.95 | s | C 1, C 3, C 4 | |||
| N 2 | NH | 12.79 | s | 3.94 | C 6, C 5 | ||
| N 3 | NH | 8.6 | t5.5 | C 10, C 9, C 11 | |||
| N 4 | NH2 | 7.45 | s | ||||
| N 5 | N |
*Calculations were performed using the HOSE code based approach.
The Molecular Connectivity Diagram (MCD) created by the program is shown in Figure 1.
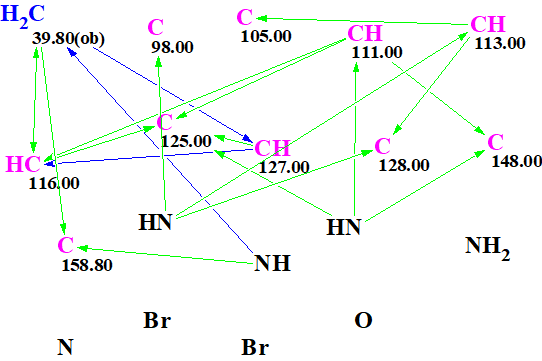
Figure 1. Molecular Connectivity Diagram (MCD).
Strict structure generation was initiated from the MCD. Only structures containing 5-6-membered cycles were allowed through in this computation. Results: k = 2736→426→157, tg = 2 s.
13C chemical shift calculations and structure ranking according to the methodology common for ACD/Structure Elucidator were carried out. The four highest ranked structures which had very low average and maximum deviations of predicted carbon chemical shifts are presented in Figure 2.
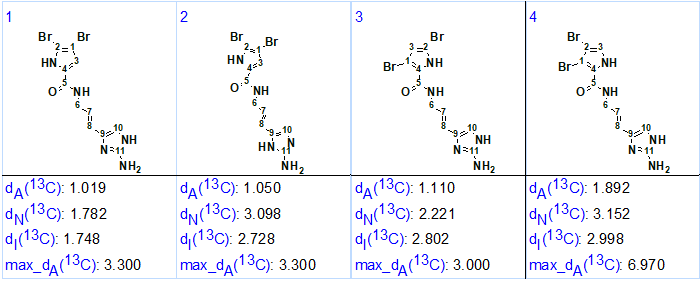
Figure 2. Four top ranked structures of the output file. The structures are ranked in increasing order of dA(13C) deviation value which corresponds to HOSE code based chemical shift prediction.
The small and close dA(13C) and max_dA(13C) values calculated for these structures prompted the utilization of DFT calculations [4] for further structure differentiation and selection of the most probable structure. Here we will present the results of 13C chemical shift calculations for competing oroidin structures as described in [5].
Figure 2 shows that three out of four predicted isomers of oroidin (#1, #3 and #4) were isomers of the bromopyrrole fragment. As the structure #2 was a tautomer of the aminoimidazole fragment it was excluded from further analysis.
DFT analysis of the three isomers suggested by ACD/Structure Elucidator (#1, #3 and #4) for oroidin were done using the SPARTAN program [6] at the EDF2/6-31G(d)//B3LYP/6-31+G(d,p) level of theory followed by empirical corrections [6]. Since the three isomers of oroidin were different only in the configuration of the dibromopyrrole fragment, chemical shift analysis was carried out only for that part of the molecule approximated with the following dibromopyrrole-N-methyl-carboxamides (Figure 3).
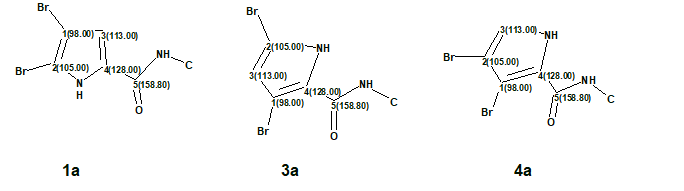
Figure 3. The molecular fragments used for DFT based 13C chemical shift prediction. Atom numbers and the corresponding experimental 13C chemical shifts assigned by the program are shown.
The results of quantum-chemical calculations are presented in Table 2.
| Carbons | Exp. | 1a | 3a | 4a |
| 1 | 98.0 | 100.1 | 97.8 | 100.2 |
| 2 | 105.0 | 102.6 | 97.9 | 103.4 |
| 3 | 113.0 | 111.4 | 117.2 | 121.0 |
| 4 | 128.0 | 127.7 | 128.8 | 126.7 |
| 5 | 159.0 | 161.4 | 161.5 | 161.2 |
| RMSD, ppm | – | 1.93 | 3.87 | 3.95 |
| max_d, ppm | – | 2.4 | 7.1 | 8.0 |
As seen in Table 2, the correct fragment structure 1a obviously has the lowest RMSD and max_d deviation of 1.93 and 2.4 ppm, respectively. Structure 3a, which had very similar carbon chemical shifts with structure #1 (refer to Figure 2) by CASE analysis with ACD/Structure Elucidator, has more than twice as high RMSD and nearly tripled max_d values of 3.87 and 7.1 ppm, respectively. It is noteworthy, that the chemical shifts of quaternary carbons bearing bromine atoms were well predicted by the empirically corrected DFT calculation carried out by SPARTAN [6].
The oroidin structure with assigned 13C chemical shifts is shown below.
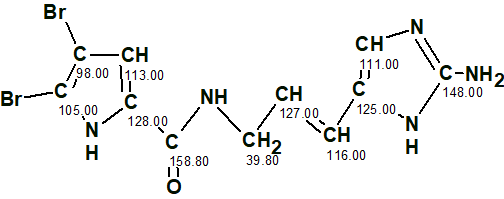
References
- M. Köck, J. Junker, T. Lindel. (1999). Impact of the 1H,15N-HMBC Experiment on the Constitutional Analysis of Alkaloids. Org. Lett., 1(13): 2041–2044. DOI: 10.1021/ol991009c
- T. Lindel, J. Junker, M. Köck. (1999). 2D-NMR-Guided Constitutional Analysis of Organic Compounds Employing the Computer Program COCON[#]. Eur. J. Org. Chem., 1999(3): 573-577. DOI: 10.1002/(SICI)1099-0690(199903)1999:3<573::AID-EJOC573>3.0.CO;2-N
- S. Rasapalli, V. Kumbam, A. N. Dhawane, J. A. Golen, C. J. Lovely, A. L.Rheingold. (2013). Org. Biomol. Chem., 11, 4133–4137. DOI:10.1039/c3ob40668g
- A. V. Buevich, M. E. Elyashberg. (2016). Synergistic combination of CASE algorithms and DFT chemical shift predictions: a powerful approach for structure elucidation, verification and revision. J. Nat. Prod., 79(12): 3105–3116.
- A.V. Buevich, M. E. Elyashberg. (2018). Towards unbiased and more versatile NMR-based structure elucidation: A powerful combination of CASE algorithms and DFT calculations. Magn. Reson. Chem., 56, 493–504. DOI: 10.1002/mrc.4645
- (2018) Spartan ’14 [computer software]. California: Wavefunction, Inc.
About the Author
