November 1, 2020
by Dimitris Argyropoulos, NMR Business Manager, ACD/Labs
Symmetric Helical Synthesis Product
This month’s problem is again a bit different from the others. It comes from the Department of Chemistry, University of Bath, UK. John Lowe had been trying to challenge Structure Elucidator and asked his colleagues at the Department for interesting problems. David Carbery gave him two synthetic samples, A and B, that had been prepared by his group for some other physicochemical studies and were thought to be impossible to elucidate by NMR [1].
The samples were isomeric, with a MW of 582.2042 and a molecular formula C38H30O6. The structures were symmetric so exactly half the signals were visible by NMR, i.e. 19 carbons. The actual spectra of A and B were different, which verified that it was two different compounds. However, the differences were only in the positions of the peaks, not in the number of signals and correlations observed. The differences were also quite small in some cases.
A series of NMR spectra were recorded for these samples, including a 1D 1H, 1D 13C, COSY, HSQC and HMBC. Already from the COSY experiment it was clear that there was something else strange: There were peaks at 6.79 ppm (dt), 7.06 ppm (td), 7.14 ppm (tt), 7.3 ppm (td) and 7.25 ppm (dt) that were coupled in the order listed, so all 5 of them formed a coupling network. These are undoubtedly all aromatic protons, but we could not think of a way that an aromatic ring or rings could have 5 non-equivalent protons.
The 1D 13C spectrum also had some very closely spaced peaks: 3 signals within 0.1 ppm at the 130.7 ppm region and another 2 within 0.06 ppm in the 127.9 ppm region. (Figure 1).
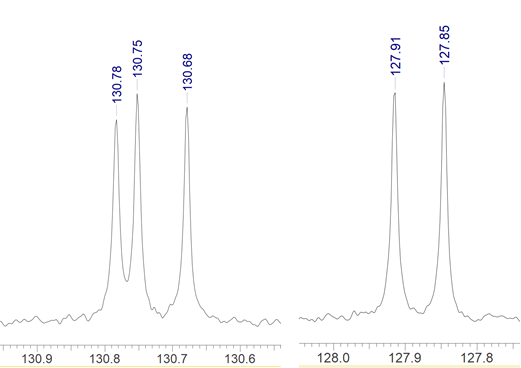
Figure 1: The regions around 130.7 and 127.9 ppm of the 1D 13C spectrum of compound A.
With such close proximity the default resolution of any 2D spectrum is not sufficient to resolve any observed correlations. This would have normally not been a problem, however in this case together with the symmetry of the molecule the difficulty of coming to a solution was increased. Structure Elucidator would need to examine every possibility for the correlations and generate multiple MCDs internally, which would increase the elucidation time. To try to resolve this problem, band-selective versions of the experiments (HMBC) were recorded, focusing on the region of 120-140 ppm in F1. Band-selective experiments use variations of the standard pulse sequences with selective instead of hard RF pulses, allowing the selective observation of a region of the spectrum. They are similar to the standard experiments in terms of acquisition time and sensitivity but since they record the same number of increments for a much narrower spectral window in F1 the resolution can be increased quite a bit. Band selective experiments are very easy to setup with modern NMR instruments and chemists should not be hesitant to use them. The result, in comparison with a conventional HMBC, is shown in Figure 2. The use of these experiments allowed the full resolution of the ambiguities and thus no dotted lines were present in the MCD.
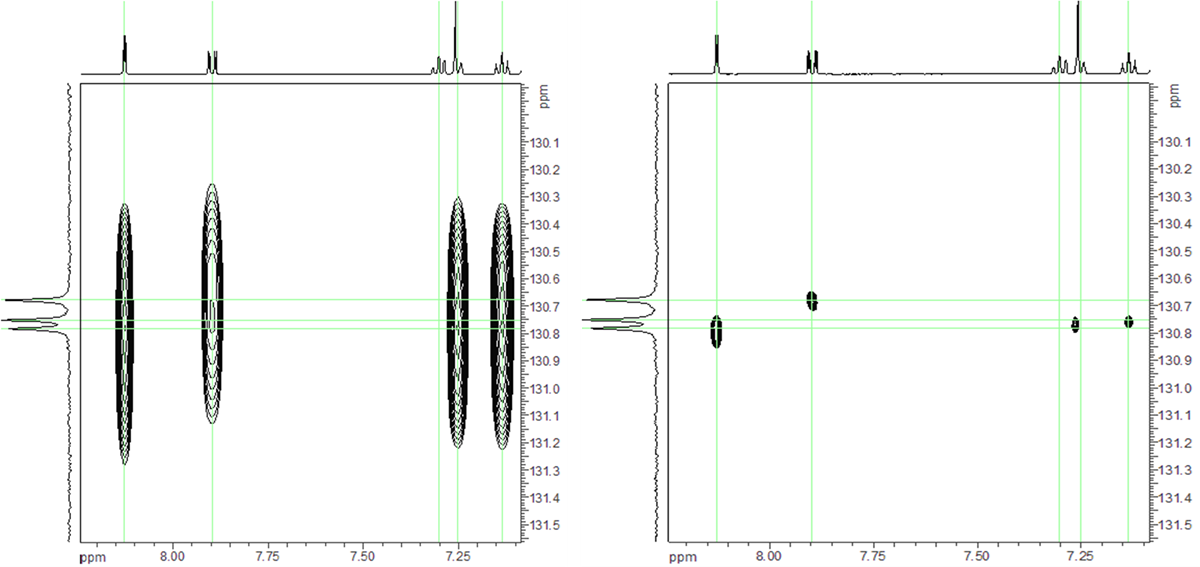
Figure 2: Conventional (left) and band-selective (right) HMBC spectrum of compound A in the region 7.0-8.0 ppm in F2 and 129.8-131.6 ppm in F1. The acquisition time for both the spectra was approximately the same, just under 60 minutes on a 500 MHz system with a standard, non-cryogenically cooled probe.
Since the compounds are slightly proton deficient and because of their symmetry, the HMBC spectra were recorded twice, optimized for two coupling constants: 8 Hz and 5 Hz. This allowed a few more correlations to be observed.
The NMR data collected for compound A are shown in Table 1. To accommodate for the symmetry of the problem the table of 13C data was edited and each signal was set to correspond to 2 carbons. The resulting MCD is shown in Figure 3.
Table 1: The NMR data extracted from the spectra and used in ACD/Structure Elucidator. Each line corresponds to 2 carbons, because of the problem symmetry.
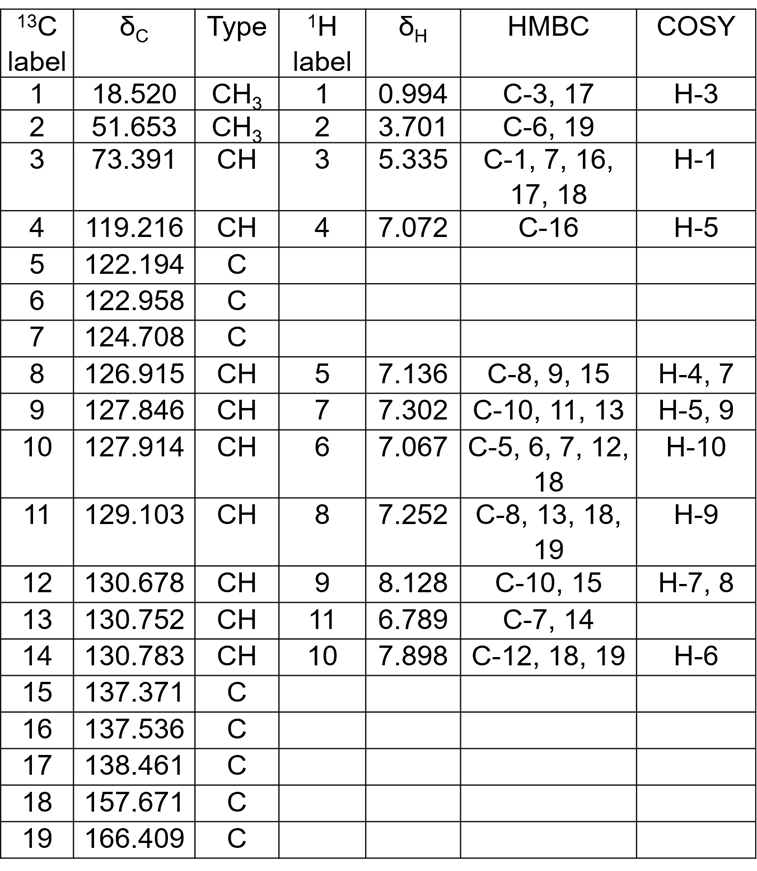
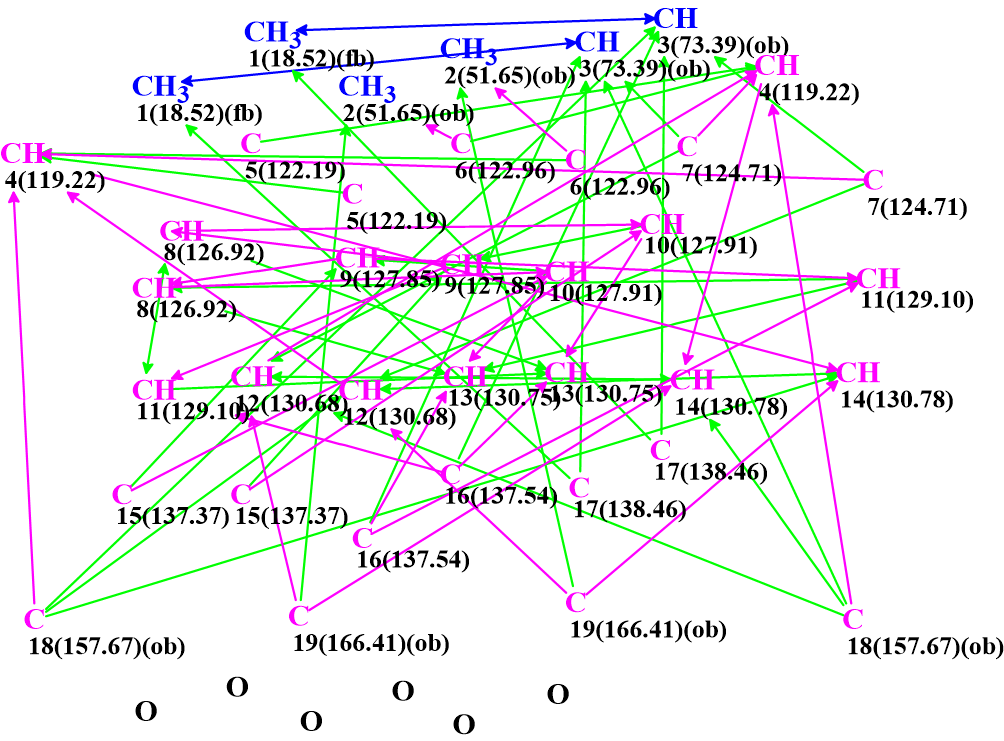
Figure 3: The MCD generated by the program, after the edits described in the text.
There are 38 carbon atoms shown in the MCD, in accordance with the MF. There are 4 sp3 and 4 sp2 atoms that have been labelled with “ob”, meaning that they must be connected to a heteroatom. The 4 sp3 ones were set like that automatically while the 4 sp2 ones manually, as they are at a chemical shift range typical for carbonyls. There are two sp3 carbons that are labelled “fb” meaning that they cannot be connected to a heteroatom. There are also 6 oxygen atoms that are “floating”, meaning that there is no evidence as to where they are bonded. We also see that some of the correlations are marked as green while others are magenta. This indicates the estimated length of the correlation: green ones are up to 3 bonds and magenta ones up to 4 bonds long. This is derived mostly by the intensity of the HMBC peaks and, when in doubt, it is set to up to 4 bonds.
There were some edits done to the MCD: The bonds between the methyl signal and the CH group were manually set as they are very clearly present based on the HSQC and COSY spectra. The hybridizations of all the other carbon signals above 119 ppm was set to be sp2 as it was certain that there are no sp carbons in the product.
Before starting the generation and since we had very high resolution HMBC spectra, the tolerance for the 13C shifts was set to 0.001 ppm instead of the default 0.0076. This helps to accelerate the elucidation as the position of each carbon signal is not allowed to vary as much.
The MCD passed all tests, no contradictions were found and no edits were made. After this strict structure generation [2,3] was initiated with the following results: k = 2449 → (spectral filtering) → 113 → (removing duplicates) → 113, tg = 1 h 46 m 26 s.
The top 3 structures generated are shown in Figure 4.
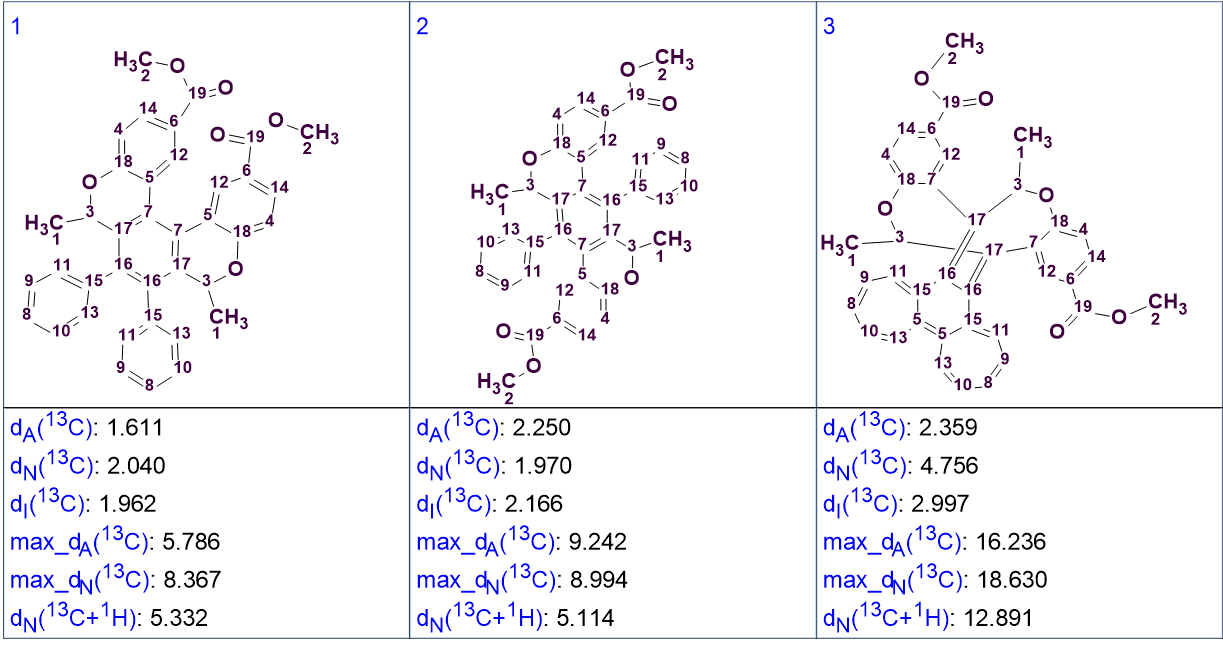
Figure 4: The top 3 structures generated by the program.
We can see that first two structures are very closely related, with the one derived from the other by “breaking” the middle aromatic ring, rotating one half, and re-forming the ring. We see that the average deviations between the predicted and experimental chemical shifts using the three methods built into ACD/SE are very close, however the maximum deviations observed for the third structure are almost double that for the other two. This, together with the highly unusual 10-membered bridged ring with double bonds effectively rules it out.
The correct structure is clearly one of the two first ones. We see that there is no clear “winner” based on the mean deviations between observed and calculated chemical shifts: Using the HOSE codes approach the first structure is correct, however using the neural networks approach the second one is better. Moreover, there is no bond correlation experiment that could have hinted at the correct one. Additionally, observing the structures more carefully, we can see the reason why we had the 5 non-equivalent, coupled aromatic protons: The mono-substituted aromatic ring is, in both cases, in a very sterically hindered environment and it is unlikely that it can rotate freely, which would have made some of its protons equivalent. So, the non-equivalency observed is because of stereochemistry reasons.
Similar results are obtained if we analyse the data for compound B. The same two structures are generated and ranked at the top, which means that the synthesis produced both isomers which were later separated and analysed. In order to solve this final problem a 2D-NOESY spectrum was recorded. This spectrum for compound B is shown in Figure 5.
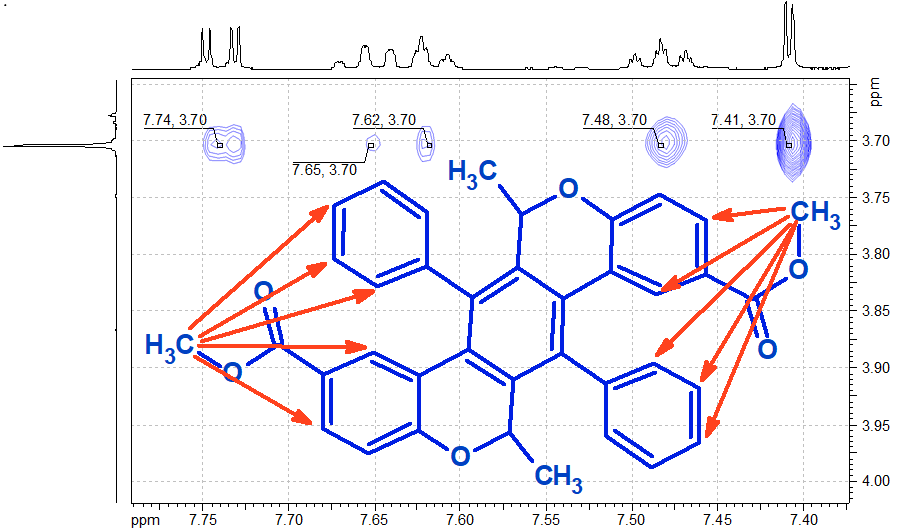
Figure 5: Expansion of the 2D-NOESY spectrum region for compound B, showing the correlations between the methyl of the ester group with the aromatic protons of the monosubstituted ring.
It is clearly seen that for the case of compound B, NOESY correlations between the methyl protons of the ester group and some of the protons of the monosubstituted ring are observed, indicating that these are close in space (ca. <5 Å). No such correlations are observed for compound A. This clearly proves that the correct structure for compound A is the first one in Figure 4, while for compound B it is the second one. It also provides a direct proof of the steric hindrance in the rotation of the monosubstituted ring: Only correlations to 3 of the 5 protons are observed, the ones that are the closest. If there was free rotation possible then correlations to all the protons would have been seen.
The final structures of compounds A and B are shown in Figure 6, together with the assigned 13C chemical shifts.
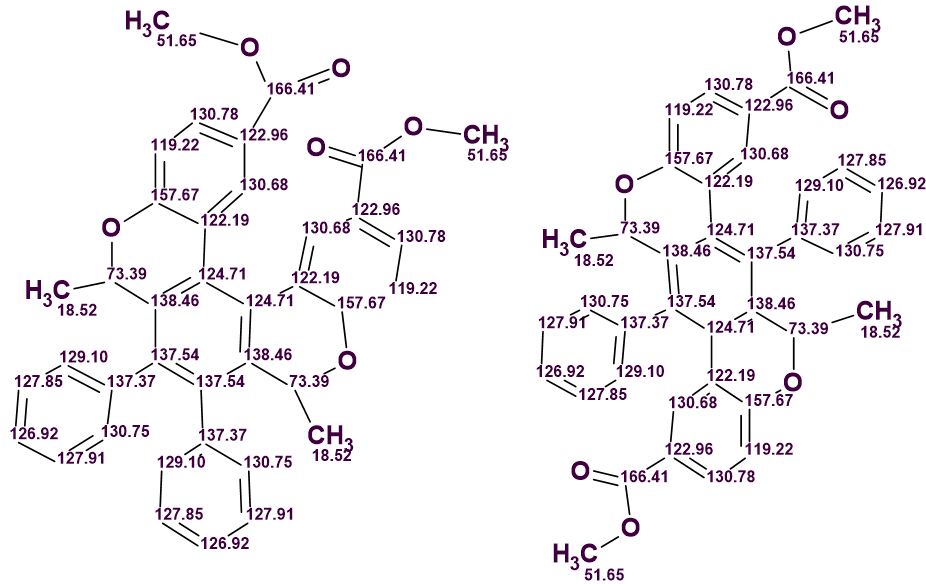
Figure 6: Elucidated structures and observed 13C chemical shifts for compounds A (left) and B (right).
In conclusion Structure Elucidator was able to solve the structures of two helical, symmetric synthetic compounds with some quite unique challenges, in a relatively short time.
References:
- Carbery, D. R.; Lowe, J. P.; Moser, A.; Argyropoulos, D., “Computer Assisted Structure Elucidation of Two Isometic, Highly Symmetrical Helical Molecules”. In EUROMAR Aarhus, Denmark, 2016.
- M. E. Elyashberg, A. J. Williams. (2015). Computer-based Structure Elucidation from Spectral Data. The Art of Solving Problems. Springer.
- M. Elyashberg, D. Argyropoulos, in eMagRes, Vol. 8 (Eds.: R. K. Harris, R. L. Wasylishen), Wiley, 2019, pp. 239.
About the Author
