June 1, 2018
by Mikhail Elyashberg, Leading Researcher, ACD/Labs
Tetrabromobenzofuropyrrole
A highly brominated compound, 2,3,5,7-tetrabromobenzofuro[3,2-b]pyrrole (1), was extracted from a marine Protobacterium, Pseudoalteromonas sp [1].
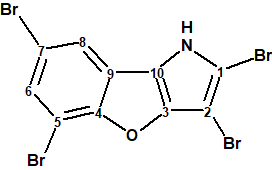
1
The extract was fractionated by reversed-phase column chromatography, followed by reversed-phase HPLC and several recrystallization steps that yielded 1.1 mg of pure compound 1. The negative mode HR-ESI-MS spectrum of 1 indicated a molecular formula of C10H379Br4NO with the monoisotopic [M – H+]– peak at m/z 467.6877 and the very characteristic isotopic pattern (2:7:10:7:2) indicative of the presence of four bromine atoms.
The structure of 1 was determined based on the high-resolution MS data, proton-proton J-coupling constant analysis, HMBC correlations and empirical carbon chemical shift predictions (ACD/CNMR Predictor v.12 was used). The molecule has only three protons and 16 skeletal atoms, which makes it highly proton-deficient, with the ratio R= n(skeleton)/n(hydrogens)= 5. Structure elucidation of such molecules is extremely challenging [2]. It is noteworthy, that only one alternative structure 2 has been examined by the authors of [1], which appears to be unsatisfactory, considering its highly proton-deficient nature:
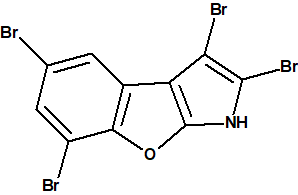
2
Spectroscopic NMR data used for elucidation of structure 1 are collected in Table 1.
Table 1. Spectroscopic 1D and 2D NMR data of 2,3,5,7-tetrabromobenzofuro[3,2-b]pyrrole.
| C Label | C Shift | C Calc Shift | XHn | H Shift | H Multiplicity | C HMBC |
| C 1 | 107.2 | 104.95 | C | |||
| C 2 | 81.7 | 84.79 | C | |||
| C 3 | 149.1 | 145.6 | C | |||
| C 4 | 155 | 153.98 | C | |||
| C 5 | 106.4 | 102.91 | C | |||
| C 6 | 129 | 134.25 | CH | 7.55 | d1.8 | C 8, C 4, C 7, C 5 |
| C 7 | 117 | 115.75 | C | |||
| C 8 | 120 | 120.49 | CH | 7.67 | d1.8 | C 4, C 6, C 7, C 9 |
| C 9 | 120.1 | 117.98 | C | |||
| C 10 | 122.6 | 119.93 | C |
These data were entered into ACD/Structure Elucidator, and the Molecular Connectivity Diagram (MCD) was automatically created by the program (Figure 1).
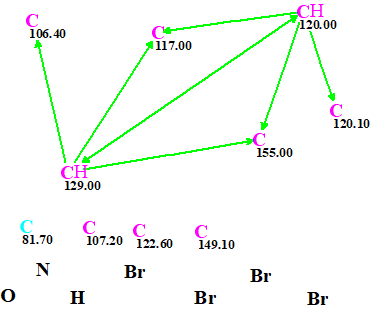
Figure 1. Molecular Connectivity Diagram. Properties of all skeletal atoms are set automatically.
MCD overview. All carbon atoms except C 81.7 received a sp2 hybridization. Hybridization of C 81.7 is allowed to be sp2 or sp3 (not sp). Due to the severe deficit of hydrogen atoms, four carbons have no HMBC correlation. This means that structure generation will be long for this small organic molecule. It also means that a large number of isomeric structures will be generated. As a side note the total number of theoretical constitutional isomers corresponding to the molecular formula C10H3Br4NO is calculated to be 9,829,354,271 (!).
Strict structure generation from the MCD accompanied with 13C chemical shift prediction and structure filtering gave the following result: k = 166,444 → 67 → 32, tg =9 m 15 s. 13C chemical shift prediction was performed for all stored structures using the three empirical methods built into ACD/Structure Elucidator (neural networks, incremental and HOSE code based) and the structural output file was ranked in increasing order of averaged deviations calculated by HOSE based approach. The six first structures of the output file are presented in Figure 2.
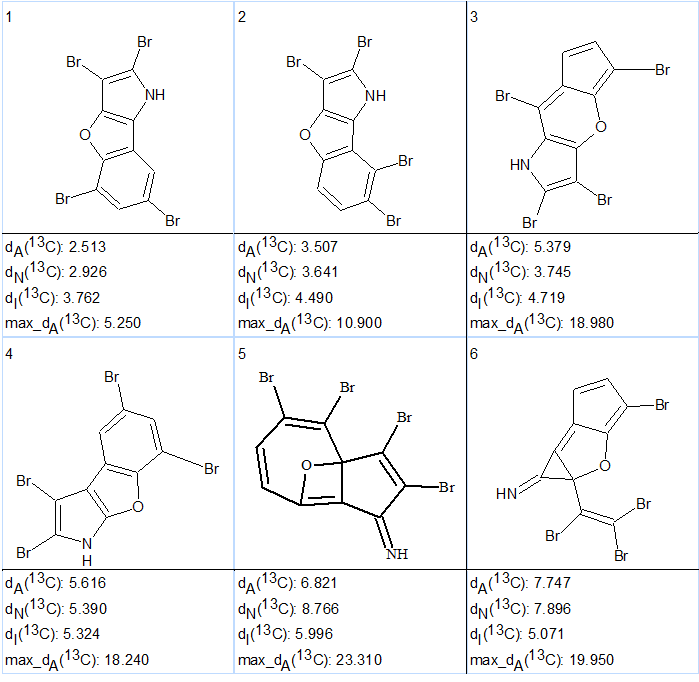
Figure 2. The six highest ranked structures suggested by ACD/Structure Elucidator for tetrabromobenzopyrrole (1).
We see that the first ranked structure coincides with structure 1, which confirms the structural assignment suggested by authors [1]. Structure of 2,3,5,7-tetrabromobenzofuro[3,2-b]pyrrole together with the automatically done 13C chemical shift assignment is shown below:
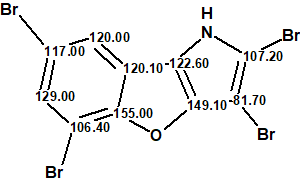
Figure 2 shows that the next (after structure #1) two structures, #2 and #3, were not consistent with a small 1.2 Hz JH,H coupling between the two aromatic protons, which supported a meta configuration of these protons. The fourth ranked structure was identical to structure 2 suggested by the authors of [1], and it has a significantly higher average and maximum chemical shift deviations, 5.6 and 18.2 ppm, respectively, compared with the 2.5 and 5.3 ppm for the first structure (1).
Even though the correct structure was determined by CASE, we have pursued the analysis by DFT to test the robustness of DFT predictions for molecules with heavy elements [3]. It has been shown earlier [4] that a synergistic combination of CASE algorithms and DFT chemical shift predictions is a powerful approach for structure elucidation, verification and revision.
DFT analysis of carbon chemical shifts of the six tetrabromobenzofuropyrrole isomers suggested by CASE was carried out at the EDF2/6-31G(d)//B3LYP/6-31+G(d,p) level of theory using the SPARTAN program [5]. Results are presented in Table 2.
| Carbons | Exp. | #1 | #2 | #3 | #4 | #5 | #6 |
| C 1 | 107.2 | 102.2 | 111.2 | 114.4 | 96.0 | 144.0 | 120.9 |
| C 2 | 81.7 | 88.3 | 95.8 | 100.0 | 93.6 | 99.6 | 105.9 |
| C 3 | 149.1 | 147.67 | 154.2 | 150.4 | 136.7 | 160.1 | 178.6 |
| C 4 | 155.0 | 152.2 | 163.2 | 149.8 | 150.8 | 173.0 | 151.6 |
| C 5 | 106.4 | 106.8 | 120.1 | 94.8 | 104.9 | 122.0 | 81.2 |
| C 6 | 129.0 | 129.7 | 133.9 | 141.6 | 128.6 | 138.4 | 158.0 |
| C 7 | 117.0 | 116.8 | 126.2 | 119.6 | 116.8 | 123.5 | 105.7 |
| C 8 | 120.0 | 118.6 | 119.6 | 108.9 | 121.2 | 125.5 | 103.7 |
| C 9 | 120.1 | 119.8 | 126.2 | 127.6 | 124.3 | 123.7 | 97.7 |
| C 10 | 122.6 | 119.2 | 130.0 | 133.9 | 107.2 | 131.0 | 149.2 |
| RMSD, ppm | – | 3.05 | 8.35 | 10.11 | 8.35 | 16.15 | 21.76 |
| max_d, ppm | – | 6.6 | 14.1 | 18.3 | 15.4 | 36.8 | 29.5 |
The table shows that the correct structure #1 (1) was also found to have the lowest RMSD and max_d of 3.05 and 6.6 ppm, respectively, among the six potential candidate structures.
Thus, the correct structure of a highly proton-deficient molecule containing four heavy atoms of bromine was found by ACD/Structure Elucidator among 9,829,354,271 theoretically possible isomers, and the structural assignment was credibly confirmed by DFT based 13C chemical shift prediction.
References
- D. Feher, R. Barlow, J. McAtee, T. K. Hemscheidt. (2010). Highly Brominated Antimicrobial Metabolites from a Marine Pseudoalteromonas sp. J. Nat. Prod., 73: 1963-1966.
- M. E. Elyashberg, A. J. Williams, Computer-based Structure Elucidation from Spectral Data. The Art of Solving Problems, Springer, Heidelberg, 2015.
- A.V. Buevich, M. E. Elyashberg. (2018). Towards unbiased and more versatile NMR-based structure elucidation: A powerful combination of CASE algorithms and DFT calculations. Magn. Reson. Chem., 56: 493–504.
- A. V. Buevich, M. E. Elyashberg. (2016). Synergistic combination of CASE algorithms and DFT chemical shift predictions: a powerful approach for structure elucidation, verification and revision. J. Nat. Prod., 79 (12): 3105–3116.
About the Author
