ACD/Labs Support Update
As we continue our integration following ACD/Labs acquisition by Revvity Signals, technical support for our software has now transitioned to Revvity Signals. Read FAQs
Are You an ACD/Labs Software User?
If your software is under maintenance, you can get answers to many of your questions in the Customer Portal (the Portal is not accessible to Freeware users or those trialing software). Register/login to the Customer Portal to access movies, quick start guides, and more resources to help you use your ACD/Labs software like a pro.
-
My download link expired. Can you re-send my download link?
You can access your download link from your account Profile. It will be under the Downloads section for 12 months from the date you downloaded it. Login to your account here to access your download link.
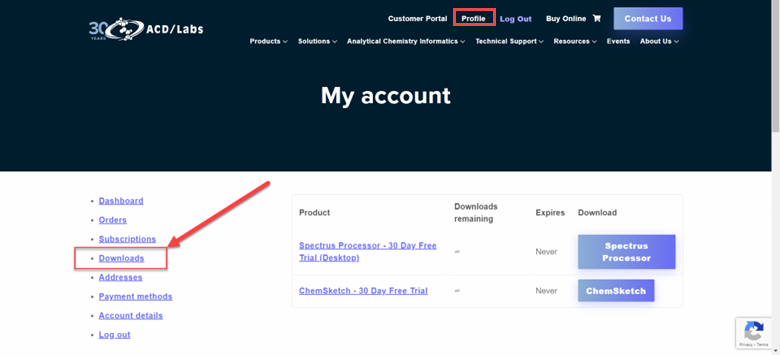
-
How do I get activation keys to install the 30-day trial download?
-
- Activation keys are automatically sent to you during the installation process. Proceed to install the software and an activation key will be sent to you later.
- If you have not received the keys after completing the installation, please follow these steps
-