New Features and Updates in V2025
Version 2025 of Spectrus Processor adds new features including expanded support for external standard qNMR, and more flexibility for integral curve positioning. There are also improvements to 19F NMR analysis, NMR and MS data import, visualization, and reporting. Read below for details, and contact us for help upgrading your software.
Highlighted Features | Improved Features | Data Import & Export | Spectrus Processor JS
Highlighted Features
Allow for Small Variations in the Integral Range of Peaks in External Standard qNMR
- You can now do external standard qNMR calculations using peaks whose integral ranges are not identical between replicates, which often occurs when working with manually defined integrals
New Parameters to Support Broader Workflows in External Standard qNMR
- You can now perform external standard qNMR calculations using replicate samples with different masses but constant volume with the help of:
- New parameters added to Concentration Calculation Tool and default report template
- Average w/w% (averaged across different replicates)
- w/w% RSD%
- Maximum Peak-to-Peak RSD% (intrasample)
- Mass and volume information included in the table of the Used Replicates popup menu
- New parameters added to Concentration Calculation Tool and default report template
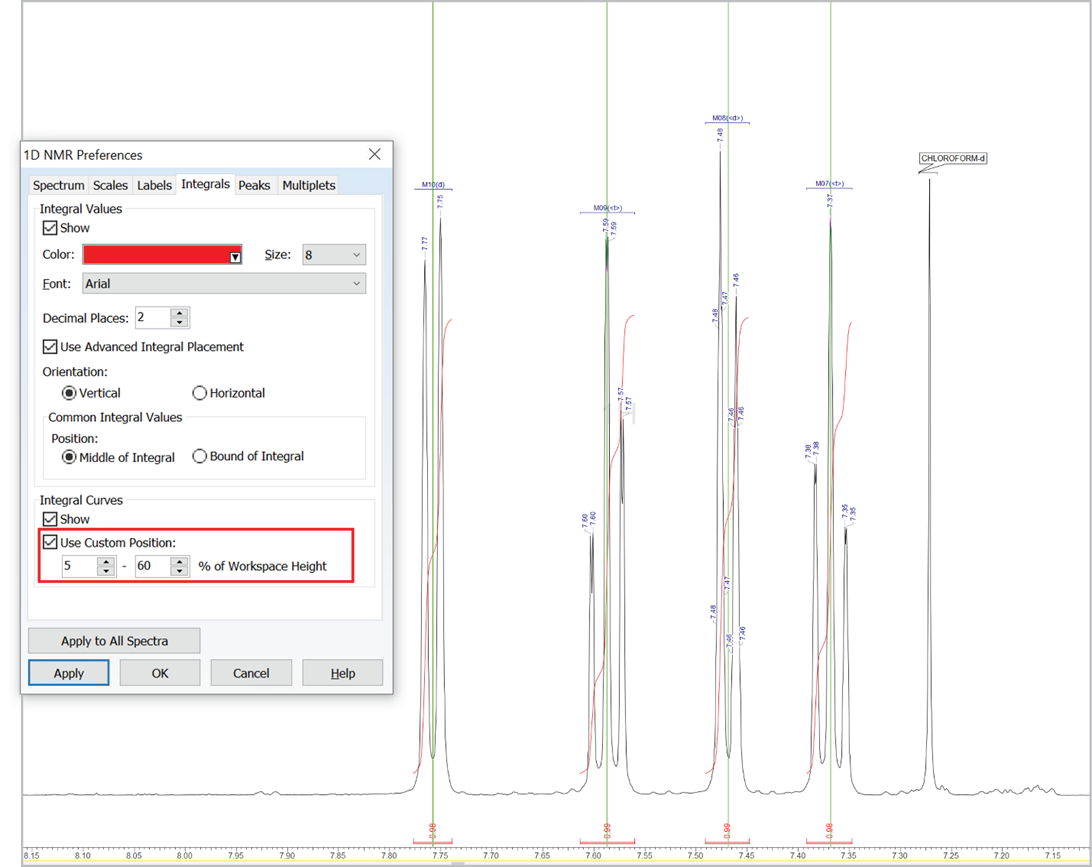
Customize the Position of Integral Curves
- You can now set a custom position for integral curves in the processing interface and reports by defining the desired vertical span of the highest curve in the displayed spectral region (as % relative to height of spectrum window, regardless of the vertical scale factor value)
Spectrus Processor JS
Highlighted Features
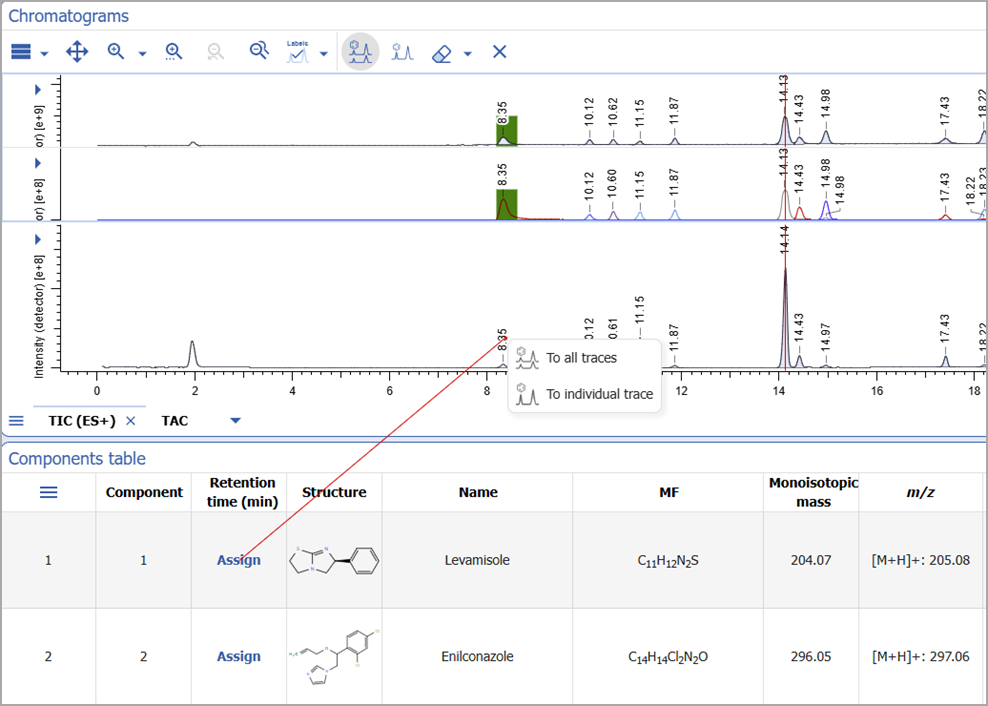
Assign Components to Individual Traces
- You now have the option to assign a component from the Components Table or Structure widget to multiple retention times across different xC/UV/MS traces
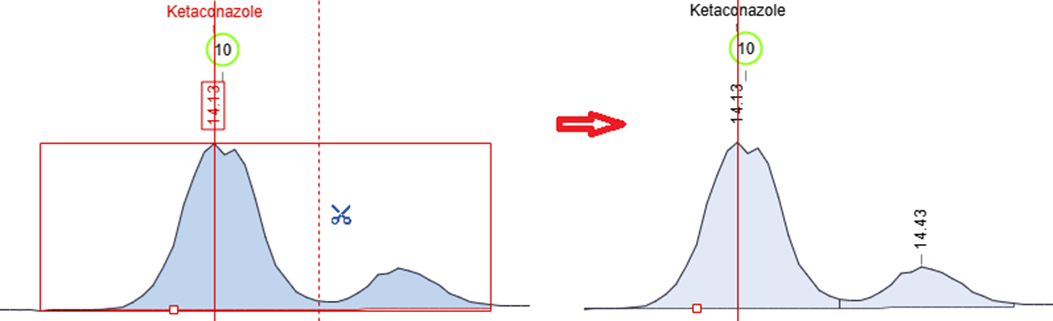
Manually Split Chromatographic Peaks
- You now have more control over peak integration with the ability to split chromatogram peaks manually using the scissors tool in manual integration mode
Improved Features
Data Analysis & Processing
Manual Assignment from Chromatograms
- You can now use the assignment tool directly from the Chromatograms widget for more straightforward analysis
- You can start the assignment from a peak on the chromatogram and complete it in the Components table or Structure widget
- Assignments can be made to all traces or to individual traces based on the selected mode
Improved xC/UV/MS Profiles
- You now have more options for setting default pre-processing parameters for the following:
- Peak detection and integration
- Mass accuracy
- Component interpretation
Improvements to Annotations for Mass Spectra and Chromatograms
- You can now more easily view and interpret overlaid mass spectra and chromatograms
- View peak labels and annotations directly on overlaid mass spectra
- Display peak labels on chromatograms in overlay mode
- Review overlaid curves with an improved interface for scan-by-scan analysis of chromatographic and MSn spectra
- Navigate datasets more easily with a redesigned curve list in the overlay layout sidebar
Improved Baseline Correction for Extracted Peaks
- You can now adjust baselines for extracted peak in manual peak integration mode, giving you more precise control over integration
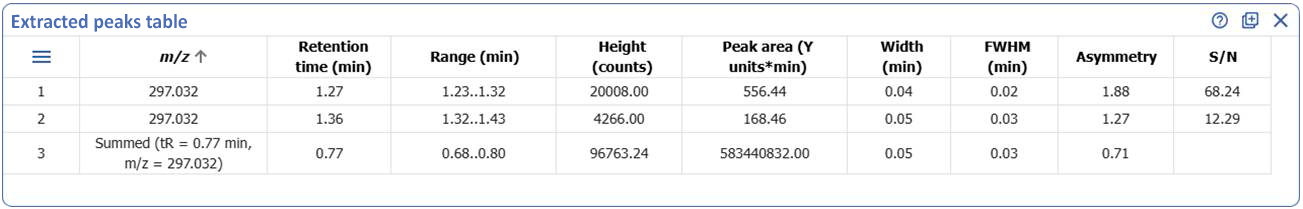
Tabulate Extracted Peaks for UV, MS, and Chromatograms
- The new ‘Extracted Peaks Table’ widget lets you clearly display and summarize peak data from individual extracted peaks directly within your dashboard
Reporting
Display MSn Component Spectra in Reports
- You now have the ability to add MSn component spectra in Pages for more comprehensive reporting
Add Extracted Peak Tables to Reports
- You can now include the new Extracted Peaks Table in both standard and template reports
- You can use the Legend to control which extracted peaks appear in the table
Ease of Use
- The eraser tool is now available for easy assignment removal of chromatographic peaks
- You now have the option to customize how MS match values are displayed:
- Optionally exclude fair match values from MS Match calculations
- Choose whether fair MS matches are displayed as color-coded in the Components table
- Control the appearance of assignment labels and previews on the chromatogram with new “color fair MS match” display options
- You can now add a new ‘Mass Values’ column to the Components table
- You now have the ability to view the XIC for any component in the Components table
Data Import & Export
Export xC/UV/MS Traces as Flat Chromatograms
- You can now export xC/UV/MS traces as flat chromatograms for simplified downstream analysis or reporting
Learn More or Buy Online
Spectrus Processor is available to purchase online, or connect with us to learn about all purchasing options.