New Features and Updates in V2025
Version 2025 of ADME Suite delivers improved accuracy for LogP and Solubility predictions through major training set expansions, enhanced support for QAF-compliant regulatory reporting, and more. Read below for details, and contact us for help upgrading your software.
Highlighted Features | Improved Features
Highlighted Features
Improved Accuracy for LogP GALAS Prediction
You can expect improved accuracy of logP calculations due to a significant expansion of the algorithm training set (Desktop and Portal Applications).
Improvements include:
- Addition of >1000 new compounds to the training set
- Addition of data from recent medicinal chemistry publications including series of compounds with novel scaffolds and varying substituents
- Expansion of coverage into the Beyond Rule-of-five (bRo5) chemical space with the inclusion of data on PROTACs, cyclic oligopeptides, and other large molecules
The linear regression scatter plots below show the comparison of experimental versus predicted logP values for newly acquired compounds, before and after the training.
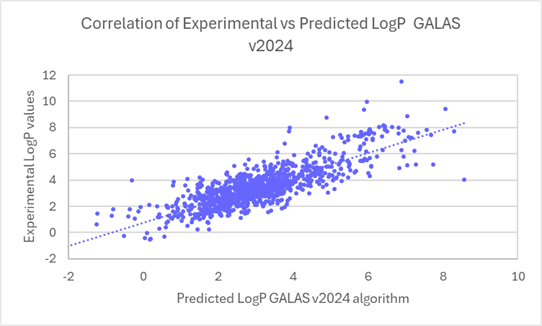
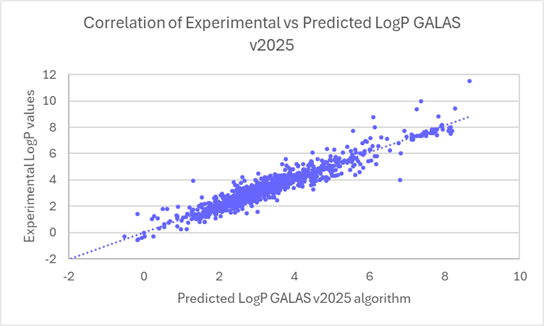
Linear regression plots demonstrate the improved correlation between experimental and predicted logP values for ~1000 compounds from v2024 to v2025.
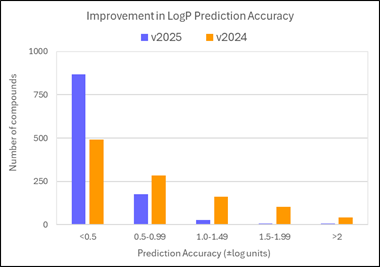
80% of logP values for these new compounds are now predicted within 0.5 log units (96% within 1 log unit). This compares to 45% within 0.5 log units (71% within 1 log unit) in v2024.
Improved Accuracy for Solubility Predictions
You can have greater trust in the Solubility calculations from a significant expansion of the GALAS algorithm training set, improving accuracy and reliability of these predictions (Desktop and Portal Applications).
Improvements include:
- Addition of >2000 new compounds to the training set (representing a 32% increase)
- Addition of data from recent medicinal chemistry publications including series of compounds with novel scaffolds and varying substituents
- Inclusion of solubilities determined from solid starting material in pH 7.4 buffer by a top pharmaceutical company (PubChem Assay ID 1159388)
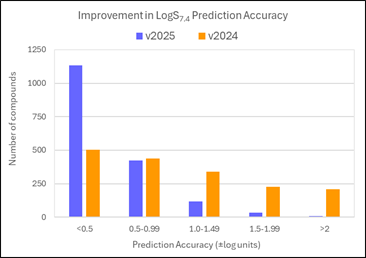
68% of LogS7.4 values for the PubChem Assay compounds are now predicted within 0.5 log units (91% within 1 log unit). This compares to 29% within 0.5 log units (55% within 1 log unit) in v2024.
Expanded Coverage of Regulatory-Targeted Reports in Accordance with QAF
You can generate fully compliant QPRF (QSAR Prediction Reporting Format) PDF reports in accordance with the QAF (QSAR Assessment Framework) initiative across multiple ADME Suite prediction modules (Desktop and Portal Applications).
- Each QPRF report is linked to corresponding QMRF (QSAR Model Reporting Format) documents that provide detailed information about the model development and validation
- All QMRFs have been updated to reflect the changes in the self-training libraries used by the models
- The following models in ADME Suite are now fully supported:
| Model Group | Model | Status |
| LogP | ACD/LogP GALAS | updated |
| ACD/LogP Classic | new | |
| ACD/LogP Consensus | new | |
| pKa | ACD/pKa Classic | new |
| Solubility | ACD/LogS0 GALAS | updated |
| BP/VP | Boiling point | new |
| Vapor pressure | new |
Improved Features in Percepta Portal
Substituent Filtering by Chemical Diversity in Structure Designer
You can now restrict the list of substituents used in analog generation to a defined number of the most chemically diverse fragments that meet your filter criteria.
- This simplifies your exploration efforts and reducing the computational workload without sacrificing the diversity of the covered chemical space
Improved Visualization of Grouped Columns
You can now easily see and quickly recognize which columns in the workspace relate to each other
- Grouped columns have color-coded stripes in their headers for easy visualization
- Multiple groups are differentiated by alternating colors for easier distinction
- You can manually group user-defined or calculated fields for more flexible data organization
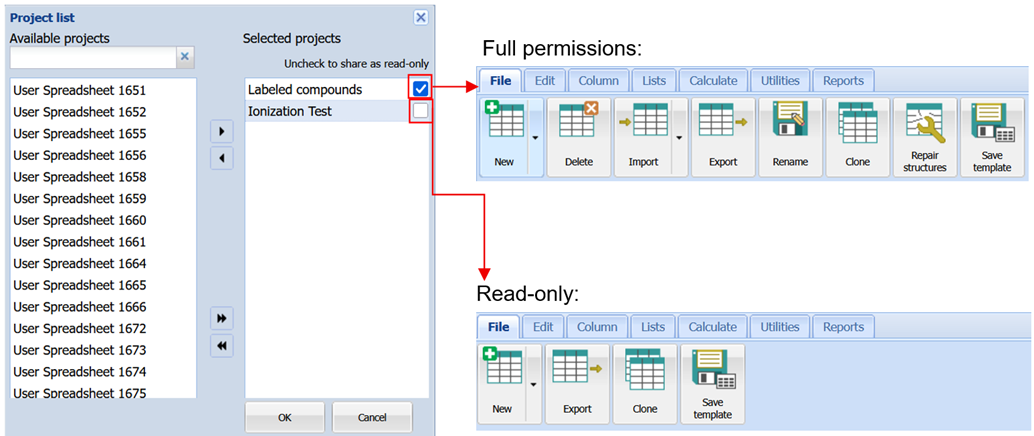
More Refined Project Access Permissions
You now have increased control over the rights granted to other team members working on a project.
- Share projects with read-only access to protect data while enabling collaboration
- Prevent accidental deletion of critical projects with a new “Protect from deletion” setting
Ease of Use
- Expect smoother spreadsheet navigation with preserved cursor position during edits
- Use full keyboard navigation in multi-value viewer/editor dialog boxes for faster input
- Avoid accidental template loss with confirmation prompts before deletion
Improved Visualization Tools Across the Plotting Workspace
You can now manage compound lists and control data visibility more consistently and efficiently across all graphical data visualization widgets:
- A unified set of commands for working with compound lists (select, save, and hide)
- Select on chart—Displays a dropdown enumerating all compound lists stored in the project and applies the selection to all compounds from the chosen list
- Save selected—Stores the set compounds currently selected on the chart as a new list
- Hide selected—Creates and loads a new temporary list with all currently selected compounds hidden from view
- Show selected—Use this option without reapplying; it now dynamically updates as selections change
- Show blanks—Always hidden if all blank records are already filtered out, providing a cleaner interface
Easier Login with OpenID Authentication Across Your Organization
- OpenID support has been expanded to integrate seamlessly with company-wide services like Microsoft Entra
- Automatically login with “Remember me” support for OpenID-authenticated users
- Automatically assign user groups for new OpenID accounts
- Benefit from improved support for secondary screens with OpenID accounts
Other Improvements
- Added support for Java 17 and Tomcat 10
ACD/Labs’ development team is eager to collaborate with organizations to improve predictions for novel compounds. Do you have accurately measured experimental values for the predictions we support? Contact us to discuss how we may work together.
Want to learn more?
Read more about the full features of ADME Suite, or contact us for help upgrading your software.