AI-Augmented Innovation & Enabling a Digital-Physical Virtuous Cycle
Innovators in various industries have recognized the value of implementing a digital-physical virtuous cycle. These innovators seek to implement this as a new foundation to support the discovery, development, and commercial launch of new products and technologies.
Specifically, this cycle refers to implementation of a continuous and mutually reinforcing loop, where digital tools and methods enhance physical processes; then, feedback from these improved physical processes informs further digital advancements.
The digital-physical virtuous cycle leads to accelerated innovation, improved efficiency, and the development of more sophisticated, data-driven solutions.
What is the DMTA Cycle?
To successfully discover and develop new therapies, innovators must perform exploratory and confirmatory experimentation. A DMTA cycle must be completed for each experiment. DMTA involves the following distinct activities:
Design
Creating a conceptual framework for potential clinical drug candidates. Design includes brainstorming, ideation, and outlining the initial specifications and functionalities of experiments. In addition to this conceptual framework for output product and physical processes, methods of analysis are carefully selected based upon the output product’s critical quality attributes and corresponding analytical target profile. Including analysis considerations as a part of the design assessment helps to assure effective decision making.
Make
Conceptual designs are transformed into physical products. Make involves synthesizing compounds. Electronic capture of observations made during activities assures that no latent variables are missed during decision-making.
Test
Experiments are subjected to pre-process, in-process, and post-process sampling (or probe-based continuous measurement). Analysis modalities include:
- Targeted Analyses—a specific sample feature or attribute is known, a suitable method to detect and measure the specific feature is available for use, and the results of the detection or measurement are unambiguous.
- Non-Targeted Analyses—a range of potential sample features or attributes is suspected, a suitable method to detect such potential features or attributes (within a specific attribute range of values) is available for use, and the results of detection or measurement is non-deterministic.
Analyze
Analysis of test results and corresponding observations made during the make step allow practitioners to derive insights and identify areas for improvement. It includes data analysis, interpretation of results, and making informed decisions for subsequent iterations.
The Vicious Cycle: Manual Data Transfer in DMTA Transitions
Each transition within the non-digitalized DMTA cycle requires significant human transposition and translation of information between systems to bridge the gap between different stages and domains of expertise.
From Design to Make
The transition from design to make requires translation of conceptual designs into logical and physical designs:
- Conceptual Design—the high-level idea and overall vision of the product. It includes abstract representations and doesn’t delve into technical specifics.
- Logical Design—the conceptual design is detailed into logical structures and relationships. It involves creating diagrams, flowcharts, and outlines that define how different components will interact.
- Physical Design—translation of the logical designs into tangible components and assemblies. It includes detailed blueprints, specifications, and instructions for manufacturing the actual product. In chemical or bioprocesses, the translation from statistics-based and stoichiometry-based process factors must be translated to physical parameters, gravimetric, and volumetric units.
In this transition, scientists and engineers must also translate statistical terms used in conceptual design into experimental and chemical terms relevant to the manufacturing process. For example, statistical models predicting product behavior must be converted into precise chemical formulations and experimental layouts.
From Make to Test
The transition from make to test involves preparing for the appropriate methods of experimental observations. This requires translating the physical designs into testable entities and defining appropriate testing protocols. Scientists must bridge the gap between the experiment or process parameters, material attributes specifications, and appropriate testing criteria. Before, during, and after each process execution, scientists must assign the appropriate sample identifiers to physical samples to ensure analysis results are associated to the appropriate process, sampling times, etc.
From Test to Analyze
The transition from test to analyze requires translation of raw test data into meaningful insights. This involves interpreting test results, identifying patterns, and making data-driven decisions. Scientists must convert the empirical data from testing into actionable feedback that informs the next cycle of design and development.
Human translation is essential in each transition of the vicious DMTA cycle, ensuring seamless communication and integration across different phases and disciplines.
Consequences of the Vicious DMTA Cycle
Ineffective transition management in DMTA leads to inefficiencies and increased risk.
 Productivity Loss
Productivity Loss
Translating from one domain to the next often requires practitioners to leverage subject matter experts to help with mapping of important, domain-specific terms from design systems, to execution systems, to analysis systems.
 Transposition/Transcription Errors
Transposition/Transcription Errors
Mapping often requires manual transposition and/or transcription of numbers and text into different software interfaces. Erroneous transposition can result in failed experiments, faulty analyses, or incorrect decisions.
Put simply, avoiding these risks across the various DMTA transitions and from digital-to-physical steps allows for a virtuous DMTA cycle.
PART I—Discovery
The Virtuous DMTA Cycle in Drug Discovery
There are a variety of specific DMTA cycles required for successful candidate nomination. These involve cross-disciplinary project teams collaborating to, ultimately, promote a reasonable drug candidate to clinical testing. Disease pathology research, biological target and pathway analysis, molecular modality selection, in-vitro assay development, in-vivo study design, and test article scale-up research all require effective DMTA cycles to matriculate through to clinical studies. Fundamentally, successful lead optimization DMTA cycles will produce the “composition of matter” which organizations claim as intellectual property in support of their ultimate launch to broad patient populations. Here, we summarize the dual-purpose of DMTA cycles during lead optimization1 in drug discovery.
AI-Enabled Drug Design—The Design Step
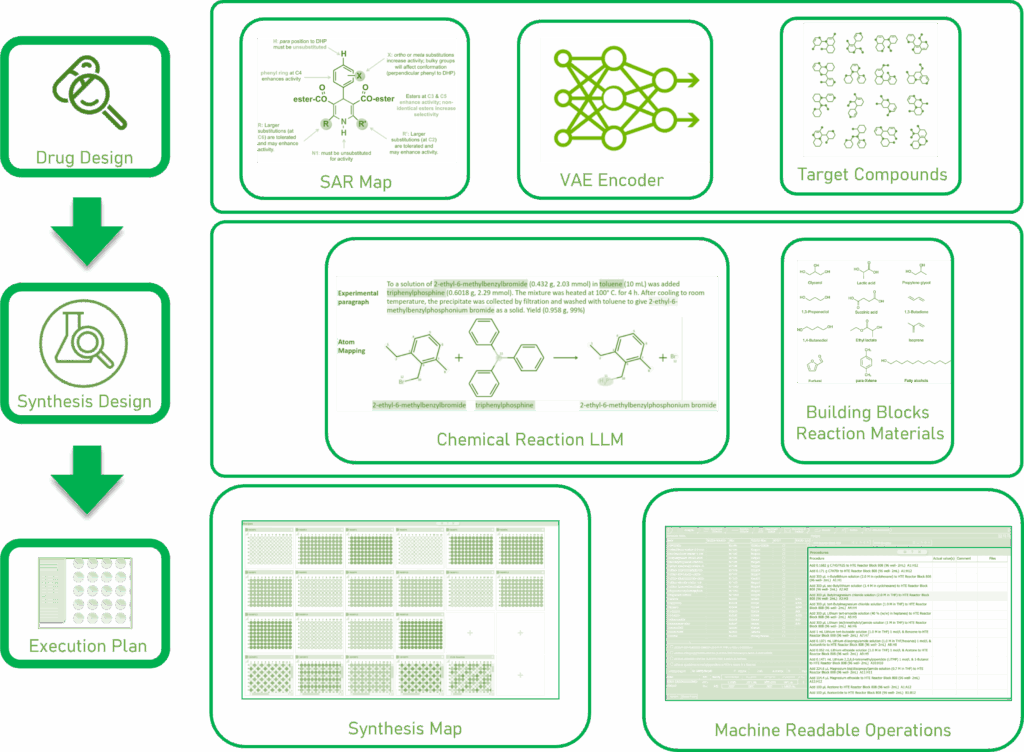
The drug design step involves the use of a SAR Map2 and a variational autoencoder to generate a set of target compounds. The synthesis design step involves the use of chemical reaction LLMs3, retrosynthesis prediction tools, and material inventory web services to generate a set of reactions. Finally, the use of specialized planning applications to support the generation of a synthesis map with machine readable operations to support the transition to digitally supported and automated execution (make, in DMTA).
The design phase of the DMTA Cycle in drug discovery addresses two key questions—what to make? and how to make it? Oftentimes innovators have solely focused on using AI to answer the first question. How AI designed test articles are synthesized, purified, and prepared for bioassay are left to manual and bespoke processes.
What to make?
Specifically, what composition of matter is most suitable for modulating the applicable drug target’s biochemical function?
The virtuous approach involves the use of generative AI tools to produce a structure activity relationships (SAR) map. These maps, for a lead series, indicate which specific atomic and functional moieties impact biological target binding, ADME, and toxicological properties. Similarly, in the case of biomolecule modalities, SAR maps indicate how specific peptide/nucleotide/
saccharide sequences and related higher order structure (HOS) functionality influences therapeutic and ADME-T properties. Once sufficient SAR information has been obtained for a lead series, medicinal chemists attempt to optimize for potency, selectivity, and overall druggability. Modern drug discovery organizations have implemented a range of generative AI4, 5 that can generate an optimized set of target compounds.
How to make it?
To confirm the success of sequential rounds of generative AI, medicinal chemists must design efficient synthetic routes for each of the target compounds. They must also minimize the total number of reactions required to produce confirmatory test articles for all AI-recommended target compounds.
Efficient approaches involve the use of well-trained retrosynthesis tools6, which perform the following design functions:
- Propose efficient, shared-path, synthetic routes
- Propose a set of reaction building blocks—available on-demand from either commercial suppliers or available within an organization’s in-house inventory
- Summarize all input materials required to execute all reaction steps for all target compounds
- Summarize reaction parameters (temperature, time, pressure, etc.)
Medicinal chemists can then order these building blocks electronically7. Ideally, these requests are fulfilled in an appropriate source plate—a ready-to-react format suitable for robotics-based operations. Furthermore, medicinal chemists use the output from retrosynthesis tools to map all requisite reactions into parallel synthesis arrays. Each array unit represents an individual reaction. The contents of each source plate (or stock locations for other input materials) are mapped to appropriate locations in a reaction container. Finally, post-reaction operations (analysis sampling, assay sample prep, etc.) are also mapped from the reaction container. The complete map of the overall experiment is contained in a synthesis map representing the complete conceptual and physical design. Medicinal chemists also use applications8 which generate an itemized, machine-friendly master procedure list that includes material dispenses, reaction operations, and sample preparation instructions. This procedure list is then disseminated to scientists to execute laboratory operations, and output as instruction files for automated robot operations.
AI-Enabled Synthesis Execution in Drug Discovery—The Make Step
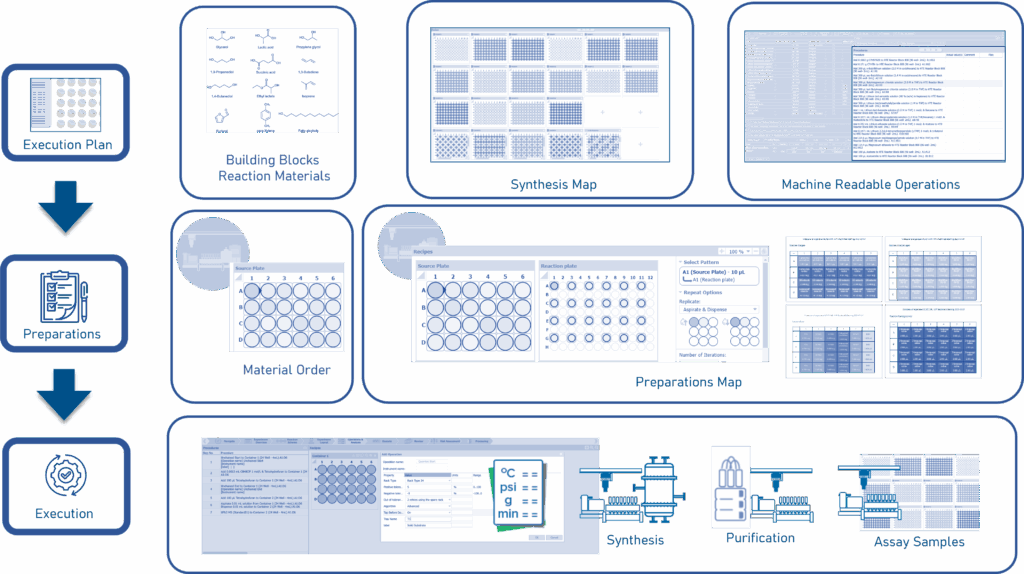
Leveraging the synthesis map and machine-readable instructions included in the execution plan, building blocks and other reaction materials are ordered and delivered in a reaction-friendly source plate format. The preparations map and corresponding operations are sent to synthesis, purification, and assay samples for execution. The digital representation of all assay samples (including applicable sample identifiers and related metadata) enables automated transition to the testing step.
Upon completion of the synthesis design, the medicinal chemist can execute each unit operation—from building block dispensing, through reaction initiation, to final workup and assay sample preparation (Figure 2). Software applications relate the materials, procedures, samples, and container locations using relational identifiers. Furthermore, physical entities can be labeled with these relational identifiers, allowing for data generated before, during, and after experimentation to be associated to the appropriate digital entities defined during the design step. Due to the modularity of modern synthesis labs, the format of machine instructions must conform to the format requirements of each device supporting each unit operation. The master procedure list is segmented by device, then electronically submitted to each device’s operating software interface. In accordance with ALCOA conformance standards9 and upon completion of each operation, device log files are associated to the applicable procedure list item(s). Any variation between planned operations and executed operations is recorded10. Downstream operations use the logfile information to account for such variation.
The output from all executed procedures results in a set of physical output materials. These materials are electronically and physically assigned to designated containers, labeled with appropriate machine-and-human-legible material identifiers. Identifiers are related to information in appropriate software applications including output material identity, material metadata, container or vessel IDs (e.g., plate and well ID).
AI-Enabled Testing in Drug Discovery—The Test Step
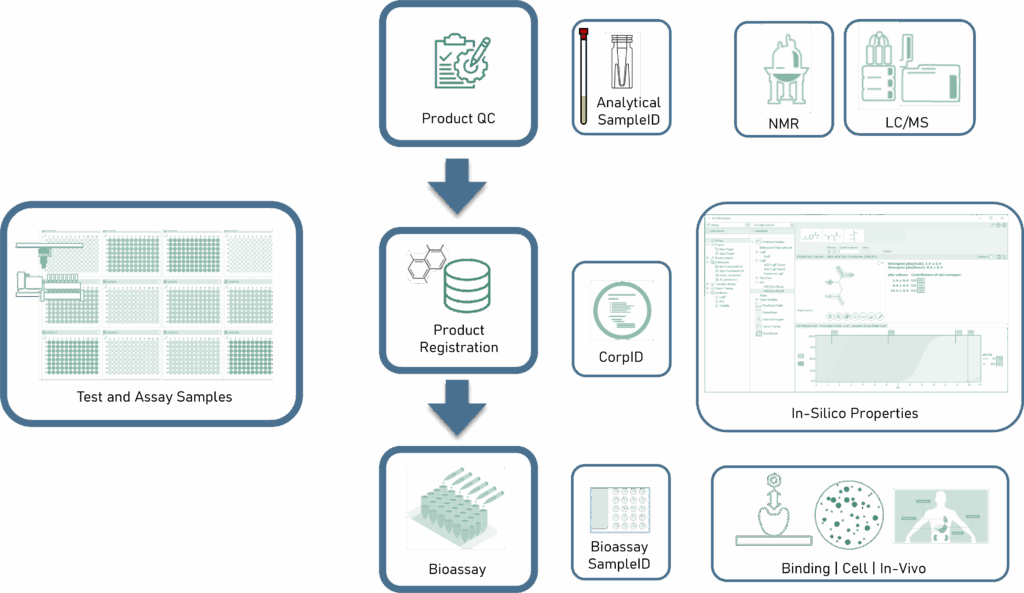
Similarly to design, the testing of output materials (i.e., purified target compounds) serves a dual purpose.
- Subjecting materials to a wide range of project-applicable bioassays to confirm the performance of the target compounds as possible (future) drugs.
- Identity and quantitative compositional testing to assure accurate SAR.11
As such, the modern DMTA cycle accounts for all applicable tests required to inform the overall lead optimization effort. Samples of output materials from reaction execution steps are prepared, then labeled with system-derived sample identifiers, including:
- Reaction quality control samples—type (preprocess, in-process, and post process) and preparation information.
- Product registration samples—including chemical identity, purity, salt stoichiometry, and any applicable physical form information that is required for assessing bioassay results for SAR map.
- Test article samples—including concentration, assay role (e.g., control, standard, blank, replicate number, etc.).
Software systems that support the overall DMTA cycle facilitate the capture and storage of sample information with identifiers. Additionally, these systems also generate instructions for scientists and robots for each sample preparation step (aspirate, dispense, dilute, etc.). This assures sample-to-data integrity and the efficient preparation of all samples via automated unit operation execution.
The testing workflow starts with generation of the complete execution of test and assay sample preparation tasks during the make step. Software systems that support the test process generate analysis equipment sequence lists, which include sample identifiers arranged in the order that the physical samples are submitted to each device’s operating queue. This allows the datasets generated by each test and assay device to be automatically associated to the appropriate material via applicable sample identifiers described above.
To assure reliable and high-quality SAR, quality control (QC) analysis is performed before bioassay. Drug discovery organizations establish minimum quality thresholds as a prerequisite to material registration. The registration step (Figure 3) requires that purified reaction product information includes identity, composition, and related physical form information. The resultant characterization information can further inform the SAR. This is especially important in cases where unexpected results are obtained during testing. In cases where a target compound’s potency is significantly different from the expected SAR, having such physical form information may reveal the source of such unexpected results.12
Upon product registration, integrated prediction systems generate a set of orthogonal structure and physical form-related properties based on data included in the registration submission (chemical structure, salt form, stoichiometry, etc.).
Table 1. Physicochemical, ADME, and toxicological descriptors generated upon product registration.
Physicochemical
- Aqueous Solubility
- Boiling Point/Vapor Pressure
- LogD
- LogP
- pKa
- Sigma
ADME
- Blood Brain Barrier Permeation
- Cytochrome P450 Inhibitors
- Cytochrome P450 Substrates
- Distribution
- Maximum Recommended Daily Dose
- Oral Bioavailability
- Passive Absorption
- P-gp Specificity
- PK Explorer
- Regioselectivity of Metabolism
Toxicity
- Acute Toxicity
- Aquatic Toxicity
- Endocrine System Disruption
- Mutagenicity
- Health Effects
- hERG Inhibition
- Irritation
AI-Enabled Analysis in Drug Discovery—The Analyze Step
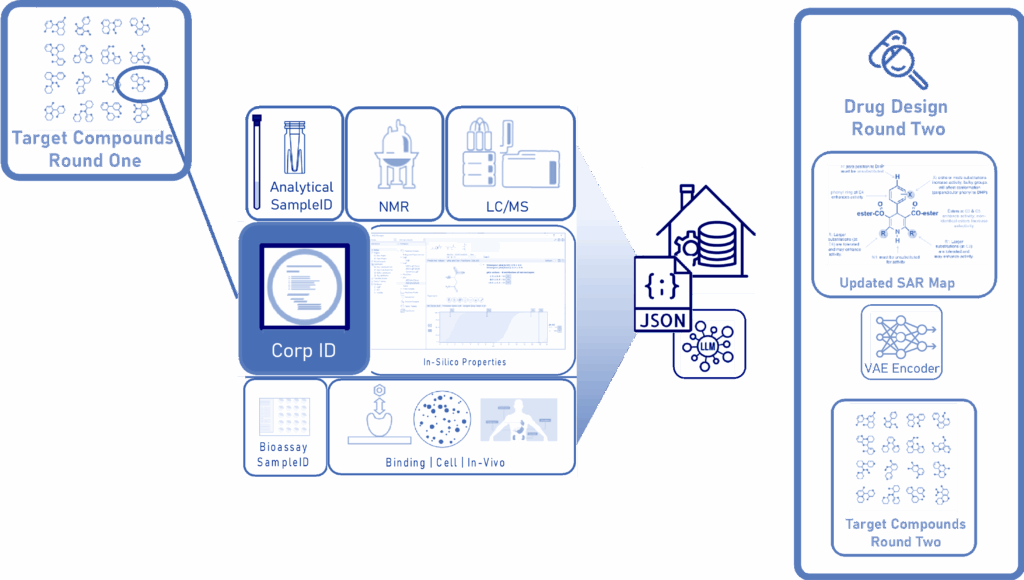
The first analysis activities support the processing and interpretation of product QC and bioassay data. Each device that is used in sample analysis produces datasets in specific (often proprietary) formats. (The variety of tools that support this primary analysis step has been described elsewhere.)13, 14, 15
Modern drug discovery organizations intend to leverage the output of these analyses by:
- Aggregating processed data into a data warehouse.
- Implementing a rigorously enforced controlled vocabulary for all bioassay and product QC Results, as well as associated metadata structured as JSON Objects.
- Relating the test results by applicable sample, corporate identifier, and in-silico properties (generated for product registration).
Scientists can then update applicable SAR maps based on the bioassay test results.
Accelerating Clinical Candidate Nomination
Enabling the modern, virtuous DMTA cycle offers significant benefits to drug discovery productivity. By leveraging LLM-based prediction models and re-training them with JSON objects generated from data analysis, organizations can continuously improve the accuracy of their predictions. This approach allows for the generation of new target compounds and facilitates a seamless and iterative process. Consequently, the integration of structured data, controlled vocabulary, and advanced analysis tools not only enhances the efficiency of the drug discovery process but also accelerates the development of innovative therapeutics. Ultimately the virtuous DMTA cycle allows project teams to optimize lead series and arrive at molecules which meet clinical candidacy specifications, faster than ever before.
PART II—Development
The Virtuous DMTA Cycle in Pharmaceutical CMC
The DMTA cycle plays a pivotal role in advancing drug development by systematically structuring scientific workflows to ensure efficiency and compliance. Two critical categories act as cornerstones of pharmaceutical innovation and quality assurance:
Process Development Studies focus on developing robust processes that adhere to corporate quality assurance standards and regulatory guidelines. These ensure that methodologies align with industry requirements for reproducibility and reliability.
Output Material Characterization Studies rigorously assess the physical, chemical, and functional attributes of reaction products, purified materials, or formulated compounds. These characterization studies are essential for verifying that materials meet predefined performance and compositional specifications, safeguarding product quality and efficacy.
Together, these encapsulate the DMTA cycle’s integrative approach to bridging scientific precision with regulatory compliance in chemistry, manufacturing, and controls (CMC) development and eventual commercial production.

Accounting for Parameters and Objectives—The Design Step
For all studies in pharmaceutical CMC development, the following approaches help assure that the complete scope of parameter and objectives are contemplated and covered.
Statistical Design of Experiments (DoE)
Statistical DoE plays a crucial role during the design phase of the DMTA cycle by enabling efficient exploration; and correlation between each critical process parameter (CPP) and the output material’s critical quality attributes (CQAs). Full factorial experimental designs require exhaustive testing of all parameter combinations and can become prohibitively resource-intensive in high-dimensional spaces. DoE, on the other hand, employs fractional factorial approaches to systematically investigate the influence of multiple factors simultaneously. This methodology not only reduces the number of experiments needed but also identifies significant factor interactions and main effects with high precision. By leveraging advanced statistical models, DoE provides a robust framework for mapping the parameter landscape and ensures that researchers can predict and optimize outcomes with minimal trial-and-error. This efficiency enhances the design’s scalability and fidelity, ultimately streamlining the transition from conceptual studies to actionable development strategies.
Bayesian Optimization
Bayesian optimization16 provides a powerful methodology for identifying optimal process parameters in CMC development and aligns closely with Quality by Design (QbD) principles. By leveraging probabilistic models (e.g. Gaussian processes) Bayesian optimization systematically explores the parameter space to predict and refine the relationship between critical process parameters (CPPs) and predefined objectives. Bayesian is particularly effective in high-dimensional spaces and scenarios where experimental trials are costly or time-consuming.
The process begins with an initial dataset of experimental results, which is used to construct a surrogate model that approximates the underlying function. This allows for effective mapping of parameters to process outcomes.
Bayesian optimization iteratively updates this model by balancing exploitation (refining known promising regions) and exploration (investigating less-certain areas). This ensures that researchers systematically converge on configurations that maximize efficiency, scalability, and reproducibility in synthetic routes.
The incorporation of predictive models enables researchers to preempt challenges, such as minimizing undesirable side reactions or optimizing reaction yield, without requiring exhaustive experimentation. By prioritizing the most informative experiments, Bayesian optimization reduces resource consumption while delivering actionable insights, laying the foundation for a reliable and compliant production strategy.
DMTA Study Summary—Critical Process Development in CMC
Table 1 summarizes the various CMC processes required to deliver suitable clinical trial materials that meet the demand of clinical trial operations.
Table 1. A summary of CMC Process Development Studies, their objectives, and how CQAs are measured.
| PROCESS TYPE | KEY PROCESS OBJECTIVES | CRITICAL QUALITY ATTRIBUTES |
| Drug Substance Synthetic Route | Optimize synthetic or biosynthetic pathways using quality by design (QbD) principles to define critical process parameters (CPPs) and objectives. | Purity, yield, potency, molecular structure consistency, reaction selectivity. |
| Drug Substance Preformulation | Characterize physical and chemical properties of the drug substance to ensure compatibility with formulation matrices. | Solubility, stability, particle size, polymorphism. |
| Drug Product Formulation | Develop and optimize the drug product formulation to ensure proper bioavailability, stability, and dosage accuracy. | Dissolution rate, assay, uniformity of dosage, shelf life. |
| Drug Substance Test Methods | Establish and validate analytical methods to test drug substance against predefined specifications. | Identity, purity, potency, impurity profile. |
| Drug Product Test Methods | Develop and validate test methods for the drug product to ensure quality and compliance. | Content uniformity, dissolution profile, sterility, appearance. |
In addition to effective study designs, Innovators within the pharmaceutical industry have implemented digitalization and AI/ML-based technologies to further assure that DMTA cycles are efficient, economical, and optimal. Moreover, these technologies allow for utilization of process development digital twins for future CMC development projects.
AI-Enabled, Drug Substance Route Development
Digitally supported, AI-augmented route development for the drug substance synthetic route includes several steps from synthesis optimization design to execution and sampling (Figure 6).
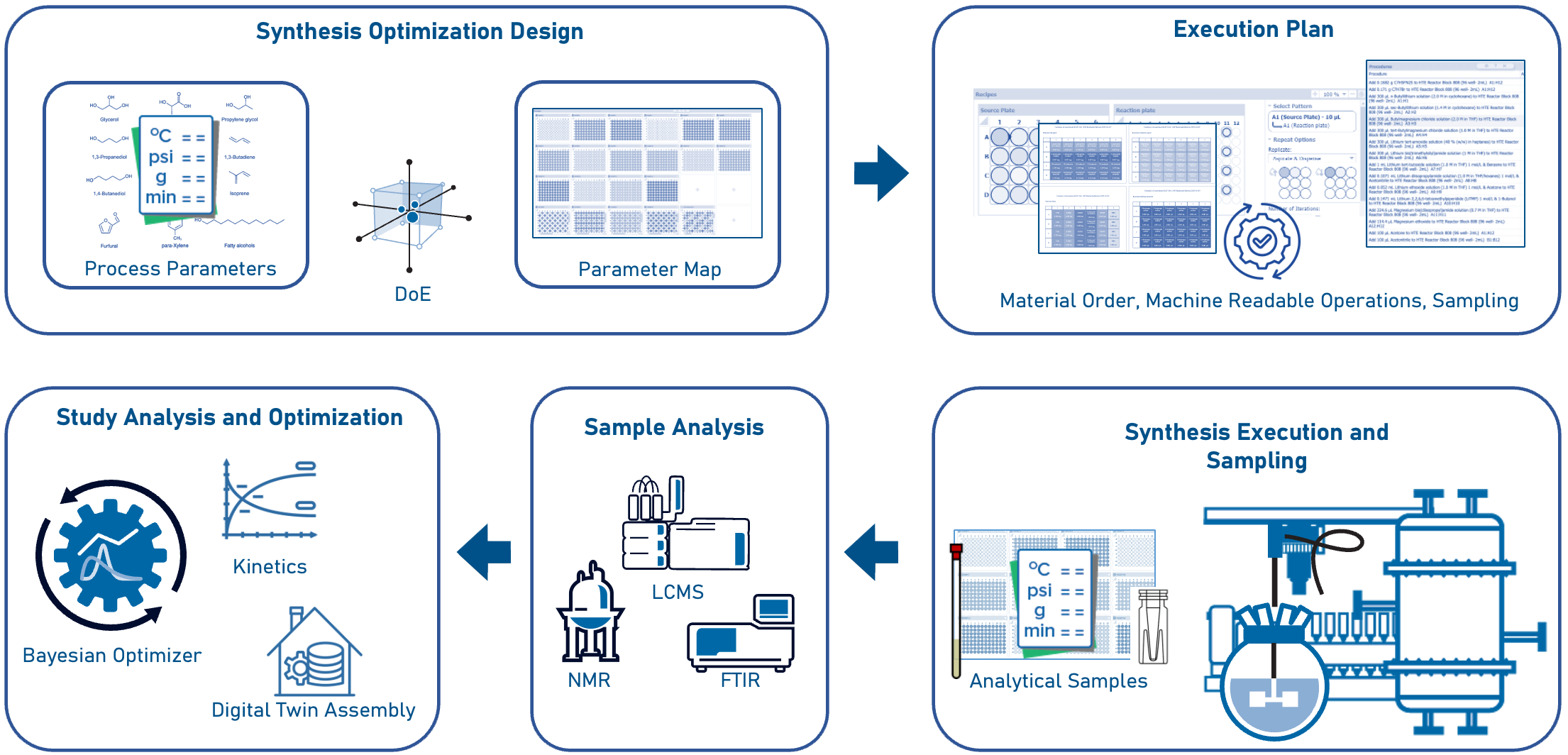
Designing the Optimal Synthetic Route
An efficient approach to designing the optimal synthesis route of a drug substance involves the use of well-trained retrosynthesis tools17 and statistical DoE tools.18 These tools perform the following functions:
- Propose efficient reaction classes for each stage of the drug substance synthetic route.
- Define arrays of reaction materials (catalysts, solvents, etc.) and related commercially available starting materials required to execute reaction stages.
- Define ranges for critical reaction parameters (temperature, time, pressure, etc.).
- Calculate efficient combinations of materials and operating parameter values, resulting in a process parameter map for the entire synthetic route.
From Conceptual Design to Physical Planning and Execution—The Make Step
Once a QbD-based parameter map has been generated, process chemists will electronically map the specified reaction trials to physical experiment designs often employing high-throughput experimentation (HTE)19 and automated reactor platforms20.
Process chemists order these materials electronically21. Ideally, these requests are fulfilled in an appropriate source plate—a ready-to-react format suitable for robotics-based operations. Furthermore, chemists will use the output from various study design methodologies to map trial experiments into parallel arrays. Each array unit represents an individual experiment trial. Contents of each source array (or stock locations for other input materials) are mapped to appropriate locations in the physical experimentation platform. Finally, post-reaction operations (analysis sampling, assay sample prep, etc.) are also mapped from the reaction container. The complete study design is represented as a digital synthesis map that contains the following:
- Conceptual design
- Physical material plan including physical and stoichiometric amounts
- Unit operations with specific operating units and times
- Sampling operations
- Experiment completion operations
Process chemists also use applications22 that generate itemized master procedure lists. These procedure lists are used:
- By scientists as concise instruction lists for manual material dispense, reaction operations, and sample preparation; and,
- As machine-friendly instruction files for automated robot operations facilitate efficient execution and in-process sampling.
Sample Identity and Purity Analysis—The Test Step
Samples of input materials, in-process materials, and post-process materials are collected at specified times in accordance with process study designs. All samples are subjected to a variety of analytical characterization studies. For drug substance routes, the extent to which a specific reaction has met process objectives can be measured as follows:
- Which experiment(s) produced the correct reaction product?
Solution-state NMR23 and ATR-FT-IR24 typically confirm product molecular identity. - Which experiments lead to optimal reaction product yield, titer, rate, and purity?
LC/MS analysis confirms the crude reaction product concentration—observed as either a relative percentage of substance-related impurities, or quantitatively (by mass percent) using appropriate reference analytical standards.25
Process Analysis using Process Digital Twins—The Analysis Step
Completion of analytical data interpretation allows for the assembly of a process digital twin, comprised of the following datasets for each experiment:
- Material and physical process parameters
- Pre-process, in-process, and post-process analysis results.
These datasets are subjected to Bayesian analysis which results in a set of recommended parameters predicted to achieve or exceed process objectives. Execution of synthesis experiments using Bayesian-recommended parameters allow scientists to investigate iterative rounds of experiments for further optimization.
Drug Substance Preformulation Studies
Upon completion of synthetic route development, similar DMTA cycles are performed to determine the optimal drug substance physical form.
Preformulation defines the physicochemical properties of the drug substance and informs formulation design.
Key studies include:
- Solubility and pKa determination via potentiometry26
- Particle size, morphology27
- Polymorphism and hygroscopicity28
- Thermal29 behavior via differential scanning calorimetry (DSC)
- Chemical stability30
Preformulation studies reveal characteristics that impact a drug product’s bioavailability, processing, and stability. These studies31 are used to determine the critical process parameters for crude reaction workup operations.
- Precipitation conditions: which produce various drug substance salt, hydrate, and solvate forms
- Wash procedure: critical for removing impurities for high-purity output material
- Crystallization conditions: impact crystallinity and particle size distributions
- Drying procedure: protocols tailored to maintain or enhance the physical properties of the output material.

Digital, AI/ML, and automation platforms support cost-effective preformulation. DMTA cycles of modern preformulation studies are conducted in the following order:
- Use QbD principles to ascertain physical form critical quality attributes and corresponding critical process parameters.
- Apply statistical DoE to establish a form parameter map which further serves as input to physical execution plans. Each resultant experiment serves to establish the significance of process parameter values set during each unit operation (precipitation, washing, reconstitution, crystallization, and drying).
- Experiment preparations, execution, and sampling operations are automatically executed by specific automation and robotics equipment. Software systems are used to build each parameter map and execution plan and to translate preparation, execution, and sampling details into digital instruction lists.
- Upon completion of all operations for all experiments, samples of input, in-process, and output materials are subjected to a series of chemical and physical characterization studies.
The following analyses help scientists determine the physical form properties of a drug substance:
- Thermogravimetric analysis (TGA)—identity and stoichiometric equivalency of solvates
- Karl Fischer titration—form hydrate stoichiometric equivalency
- Elemental analysis—trace heavy metals and salt form stoichiometry
- X-ray powder diffractometry (XRPD)—particle morphology and crystallinity
- Differential scanning calorimetry—melting point and glass transitions.
Analysis samples are digitally labeled with experiment–sample identifiers to establish the digital provenance of the originating experiment and sample collection time.
A master material digital twin is assembled for each experiment, comprising the following:
- Material and physical process parameters.
- Mapping between treatment conditions and observed physical form attributes for output materials
- Comparison of pre-process, in-process, and post-process analysis results can reveal how forms interact and, sometimes, interconvert.
Once the form mapping analysis is complete, project teams determine which form is most suitable for target formulation. The final process parameters for drug substance workup operations are determined by executing iterative optimization studies, similar to the synthesis optimization. These optimization studies result in a validation-ready process and ideal drug substance material properties. The following studies summarize the
Drug Substance Characterization Studies
Based on the wash and crystallization conditions exposed by QbD, drug substance characterization studies help to identify drug substance form(s) that exhibit the most suitable properties for product formulation.

Drug Product Formulation Studies
The formulation32 of a drug product is a meticulous process that involves selecting appropriate excipients to ensure the desired therapeutic efficacy, stability, manufacturability, and patient compliance. The decision-making processes around formulation include several critical steps:
- Excipient selection:33 excipient(s) choice is based on functionality, compatibility with drug substance, and impact on the drug’s bioavailability and stability
- Compatibility assessment: evaluation of potential interactions between the active pharmaceutical ingredient (API) and excipients using analytical techniques
- Prototype formulation development: creation of initial formulations to assess the performance and stability of the drug product
- Analytical and performance testing: study of dissolution, hardness, and content uniformity to ensure the formulation meets the desired specifications
- Stability studies: assessment of the formulation’s stability under various environmental conditions to determine self-life
- Specification setting: establishing quality specifications for the final product based on analytical data and regulatory guidelines
- Scale-up and process validation: ensuring consistent product quality in the transition from laboratory-scale to commercial-scale production.
A comprehensive overview by Pharmaceutical Technology34 summarizes the formulation development process in detail. Similar to drug substance methodologies, the use of digital systems to support the DMTA cycles for identification of optimal drug product attributes are also required. Applicable attributes include: shelf stability, active ingredient dosage consistency, dissolution rate consistency, and microbial stability.
Table 2. Pharmaceutical formulation excipient types, function, examples, and dependencies.
| EXCIPIENT TYPE | FUNCTION | COMMON EXAMPLES | API DEPENDENCY |
| Diluents (fillers) | Add bulk to tablet or capsule, especially for low-dose APIs | Lactose, Microcrystalline cellulose (MCC) | Dose strength, compressibility |
| Binders | Promote adhesion of powder particles to form cohesive tablets | Povidone, HPMC (Hydroxypropyl Methylcellulose) | Particle size, cohesiveness |
| Disintegrants | Facilitate breakup of the tablet upon ingestion for drug release | Sodium starch glycolate, Croscarmellose sodium | Solubility of API, desired release rate |
| Lubricants | Reduce friction between powder and equipment, prevent sticking | Magnesium stearate, Stearic acid | Flowability, sensitivity to compression/shear |
| Glidants | Improve powder flow during tablet or capsule manufacturing | Colloidal silicon dioxide, talc | Particle size distribution, moisture sensitivity |
| Stabilizers | Protect API from degradation (oxidation, hydrolysis, microbial growth) | Ascorbic acid (antioxidant), parabens (preservatives) | Chemical stability, oxidation/microbial sensitivity |
| Solubilizers | Enhance solubility and bioavailability of poorly soluble APIs | Cyclodextrins, polysorbates (e.g., Tween 80) | Aqueous solubility, bioavailability requirements |
| Taste-masking agents | Improve palatability of the drug product | Sucralose, flavorings (e.g., mint, fruit essences) | API bitterness, pediatric or geriatric patient compliance needs |
After Optimization DMTA—The Process Lifecycle

DMTA cycles continue after process development and optimization is complete. The lifecycle of an optimized process then transitions into downstream phases that ensure its readiness and compliance for commercial use and application in current good manufacturing practice (cGMP) laboratories. The technologies and systems employed for process optimization described above can also be utilized for the downstream steps in the overall process lifecycle.
These phases include:
- Process validation studies
- Technology transfer procedures
- Process qualification
- Final process acceptance
Together, virtuous matriculation through these lifecycle phases enables the transition of an optimized process into a fully validated, transferable, and operational methodology. This is essential for pharmaceutical development productivity and regulatory compliance.
Value Realization of Digitalized DMTA
The output from virtuous DMTA cycles result in JSON-object-based process and material digital twins. Machine learning (ML) and large language model (LLM)-based prediction and simulation tools can be automatically re-trained with these JSON objects. Without effective digital systems, data scientists spend up to 80% of their time on preparing predictive models. Data preparation time is effectively reduced to zero with digital twins and virtuous DMTA cycles. Digital DMTA systems can be used to generate more efficient process development cycles, drug substances and drug products with ideal properties, and robust, easily transferable process control methods.
Download Whitepaper
-
References (click to expand)
[1] Barcelos, M.P. et al. (2022). Lead Optimization in Drug Discovery. In: Taft, C.A., de Lazaro, S.R. (Eds), Research Topics in Bioactivity, Environment and Energy. Engineering Materials. Springer, Cham. https://doi.org/10.1007/978-3-031-07622-0_19
[2] Frey, K. (2020). Structure activity relationship (SAR) maps. Curr. Pharm. Teach. Learn., 12(3), 339-346. https://doi.org/10.1016/j.cptl.2019.12.014
[3] Vangala, S.R., et al. (2024). Suitability of large language models for extraction of high-quality chemical reaction dataset from patent literature. J. Cheminform., 16, 131. https://doi.org/10.1186/s13321-024-00928-8
[4] S. Reddy N, et al. (2024, June 30). Leveraging Latent Evolutionary Optimization for Targeted Molecule Generation. 2024 IEEE Congress on Evolutionary Computation (CEC), Yokohama, Japan. https://ieeexplore.ieee.org/document/10611790
[5] Ochiai, T., et al. (2023). Variational autoencoder-based chemical latent space for large molecular structures with 3D complexity. Commun Chem, 6, 249. https://www.nature.com/articles/s42004-023-01054-6
[6] Discovery at Your Fingertips | SYNTHIA™ Retrosynthesis Software. Synthia Online. Retrieved Sept. 15, 2025, from https://www.synthiaonline.com/
[7] Modern inventory management systems allow for orders to be fulfilled from either in-house inventory or from external suppliers from integrated data sources. NRC (US). Prudent Practices in the Laboratory: Handling and Management of Chemical Hazards: Updated Ver. Washington (DC): NAP (US); 2011. https://www.ncbi.nlm.nih.gov/books/NBK55878/
[8] High Throughput Experimentation Software | Katalyst D2D®. Advanced Chemistry Development, Inc. (ACD/Labs). Retrieved Sept. 15, 2025, from www.acdlabs.com/katalystd2d
[9] Durivage, M. (2019, Jan. 12). ALCOA Principles: Data Integrity for the FDA Regulated Industry. Quality Systems Compliance LLC. https://qscompliance.com/wp-content/uploads/2019/01/ALCOA-Principles.pdf
[10] For laboratory operations executed by scientists, applicable software interfaces allow for contemporaneous observations and notes to be recorded by the scientist.
[11] Organizations will also subject output materials to such analyses in defense of composition of matter claims in IP Applications. Pathak, S.C. (2021, Aug. 19). Basics of claim drafting for utility patent applications. Invention-Con 2021. https://www.uspto.gov/sites/default/files/documents/InventionCon2021WhatsinaPatentClaimWorkshopFinalstakeholders.pdf
[12] Bouchot, P., et al. (2020). Determination of the stoichiometry between a drug and its counter-ion by supercritical fluid chromatography using ultra-violet and evaporative light scattering detections: Application to ondansetron hydrochloride. Talanta, 218. https://doi.org/10.1016/j.talanta.2020.121166
[13] Mercer, K. (2023, Oct. 24). Enabling End-User HTE at GSK [Online presentation]. ACD/Labs Driving Efficiency with Spectrus Symposium. https://www.acdlabs.com/resource/enabling-end-user-hte-at-gsk/
[14] Standardization of Analytical Data: Best Practices. (2025, Jan. 6). ACD/Labs. https://www.acdlabs.com/resource/standardization-of-analytical-data-best-practices/
[15] FAIR Annotation of Bioassay Metadata. Pistoia Alliance. Retrieved Apr. 22, 2025 from https://fairtoolkit.pistoiaalliance.org/use-cases/fair-annotation-of-bioassay-metadata/
[16] Harrell Jr., F. E. and LaVange, L. (2024, Apr. 27). Why a Bayesian Approach to Drug Development and Evaluation? Frank Harrell Consulting. https://hbiostat.org/doc/bayes/whybayes.pdf
[17] Discovery at Your Fingertips | SYNTHIA™ Retrosynthesis Software. Synthia Online. Retrieved Sept. 15, 2025, from https://www.synthiaonline.com/
[18] Design of Experiments | JMP. JMP Statistical Discovery. Retrieved Sept. 15, 2025, from https://www.jmp.com/en/software/capabilities/design-of-experiments
[19] Big Kahuna – The Ultimate Configurable Automation Solution | Unchained. Unchained Labs. Retrieved Sept. 15, 2025, from https://www.unchainedlabs.com/big-kahuna/
[20] Automated Lab Reactors, In-Situ Analysis and Modeling Software. Mettler Toledo. Retrieved Sept. 15, 2025, from https://www.mt.com/us/en/home/products/L1_AutochemProducts.html
[21] National Research Council (US) Committee on Prudent Practices in the Laboratory. Prudent Practices in the Laboratory: Handling and Management of Chemical Hazards: Updated Version. (National Academies Press, 2011) (https://www.ncbi.nlm.nih.gov/books/NBK55868/
[22] High Throughput Experimentation Software | Katalyst D2D®. Advanced Chemistry Development, Inc. (ACD/Labs). Retrieved Sept. 15, 2025, from www.acdlabs.com/katalystd2d
[23] Emwas A-H. (2020). NMR as a “Gold Standard” Method in Drug Design and Discovery. Molecules, 25(20), 4597. https://pmc.ncbi.nlm.nih.gov/articles/PMC7594251/
[24] Alkhuder K. (2022). Attenuated total reflection-Fourier transform infrared spectroscopy: a universal analytical technique with promising applications in forensic analyses. Int. J. Legal Med., 136(6), 1717-1736. https://pmc.ncbi.nlm.nih.gov/articles/PMC9436726/
[25] Reference Materials. MilliporeSigma. Retrieved Sept. 15, 2025, from https://www.sigmaaldrich.com/US/en/products/analytical-chemistry/reference-materials
[26] Wiedenbeck E. et. Al. (2020). Potentiometric Titration Method for the Determination of Solubility Limits and pKa Values of Weak Organic Acids in Water. Analytical Chemistry, 92(14) 9511-9515. https://pubs.acs.org/doi/10.1021/acs.analchem.0c00247
[27] US Pharmacopeia, Harmonized Standards. (2016, Nov.). Particle size distribution estimation by analytical sieving. https://www.usp.org/sites/default/files/usp/document/harmonization/gen-chapter/g01_pf_30_6_2004.pdf
[28] US Pharmacopeia, Harmonized Standards. (2022, May). Characterization of crystalline and partially crystalline solids by x-ray powder diffraction (XRPD). https://www.usp.org/sites/default/files/usp/document/harmonization/harmonization-april-2021-m99730.pdf
[29] US Pharmacopeia, Harmonized Standards. (2014, Jun.). Thermal Analysis. https://www.usp.org/harmonization-standards/pdg/general-chapters/thermal-analysis
[30] Harris, J. (2022, Apr. 18). What are Forced Degradation Studies? An Introduction to Pharmaceutical Stress Testing. Advanced Chemistry Development, Inc. (ACD/Labs). https://www.acdlabs.com/blog/what-are-forced-degradation-studies-an-introduction-to-pharmaceutical-stress-testing/
[31] ICH Expert Working Group. (2009, Aug.). ICH Harmonised Tripartite Guideline | Pharmaceutical Development Q8(R2). https://database.ich.org/sites/default/files/Q8_R2_Guideline.pdf
[32] Inside drug development: The fundamental principles of pharmaceutical formulation. (2023, Nov. 10). Patheon Pharma Services. https://www.patheon.com/us/en/insights-resources/blog/inside-pharmaceutical-formulation-development.html
[33] Makkad S. et. al. (2025). Pharmaceutical Excipients: Functions, Selection Criteria, and Emerging Trends. Int J Pharm. Investigation, 15(2), 361-376. https://jpionline.org/10.5530/ijpi.20251676
[34] Hwang R.-C. and Kowalski D. L. (2005). Design of Experiments for Formulation Development. Pharm Tech, 7. https://www.pharmtech.com/view/design-experiments-formulation-development-0