May 9, 2025
by Bara Townsend, Marketing Communications Specialist
A Shifting Therapeutic Landscape
Drug discovery has gone through a remarkable diversification in the last few decades, expanding far beyond traditional small molecules to include a wide array of novel drug modalities—antibody-drug conjugates, gene and cell therapies, nucleotide-based drugs, peptides, and more—reshaping how we think about treating disease.
While these modalities hold great promise, they also bring significant challenges in manufacturing, delivery challenges, scalability, safety, and regulatory uncertainty. These limitations highlight the continued value of drug-like chemical properties such as oral bioavailability, scalability, and intracellular access—strengths where complex small molecule therapeutics often excel.
As a result, the future of medicine is being shaped by hybrid approaches that blend the precision of biologics with the flexibility and scalability of chemical-based drugs. From protein degraders to macrocyclic peptides, this new wave of complex, often large molecules shows how biology-driven design and chemical innovation can work together. And this new approach to drug design demands smarter, and accurate predictive tools to support it.
The Evolution of Small Molecules in Drug Design
Although viewed as a ‘traditional’ approach in drug design, small molecules are undergoing their own revolution, with more innovative and creative drug candidates and targeting pathways emerging over the last few years.
Advanced molecules that don’t strictly follow Lipinski’s Rule of 5 (Ro5)—larger, more flexible, and often more polar—are increasingly viable as therapeutic modalities. These compounds include:
- Protein degraders such as PROTACs and molecular glues
- Macrocyclic and constrained peptides with improved stability and target specificity
- Covalent inhibitors capable of modulating previously “undruggable” proteins
- Bifunctional and conjugated compounds designed for multifunctional activity
Despite these modalities often expanding into the beyond Rule of 5 (bRo5) chemical space, they have shown significant promise in achieving oral bioavailability, intracellular activity, and targeting challenging protein classes.
These innovations are transforming how we think about small molecules in drug design, pushing the reach of small molecules well beyond what was previously thought chemically accessible in disease treatment.
Predictive Property Tools for Novel Drug Modalities
As small molecule drug discovery advances into beyond Rule of 5 chemical space, researchers need predictive tools that can handle the structural complexity, flexibility, and size of modern therapeutic modalities, such as PROTACs and macrocyclic peptides. These often fall outside the scope of traditional predictive models trained on small, lipophilic compounds. These new generation predictive tools must go beyond classical Rule of 5 limits and support more nuanced, property-driven design goals. ACD/Labs’ Percepta® Platform—specifically the Structure Design Engine™—offers a flexible, data-driven solution for early-stage optimization of drug-like molecules, helping chemists navigate complex property trade-offs and prioritize lead compounds more effectively.
Supporting Rule of 5 and Beyond
The Structure Design Engine enables medicinal chemists to test structural hypotheses before synthesis, explore alternative analogs based on property goals, and prioritize the most promising candidates based on full physicochemical and ADME-Tox profiles. It supports both traditional small molecule workflows and the growing need for bRo5 strategies, by allowing users to tailor compound ranking criteria to their therapeutic context and needs.
Researchers can define property-based optimization strategies—such as improving CNS penetration or balancing solubility and permeability—by setting target ranges using intuitive sliders and visual indicators. The software allows precise control over fragment selection; filtering suggestions based on ionization states, sterics, or electronic effects; or similarity to known motifs. This modularity ensures that the optimization process is driven by project-specific requirements, rather than fixed thresholds.
This flexibility also extends to compound ranking through the Percepta ADMET Profiler™ module, which classifies predicted property values into favorable or unfavorable categories, enabling compound ranking based on drug-likeness. A dedicated “Lipinski” category applies the standard Ro5 criteria, assigning compounds as “good,” “average,” or “bad” depending on the number of Ro5 violations.
Alongside this, the platform also includes a similar but more flexible “Lead-like” category. Originally parameterized using traditional drug-likeness rules, this category can be customized to reflect the specific property thresholds relevant to the chemical space explored in a given drug discovery project.
For example, in a recent publication,1 researchers proposed significantly different upper limits of physicochemical properties for bRo5 compounds, and specifically for PROTACs. As illustrated in the figure below, users can easily adjust the “Lead-like” category to apply these tailored rules to align with their project’s goals:

Lipinski Ro5:
2 violations
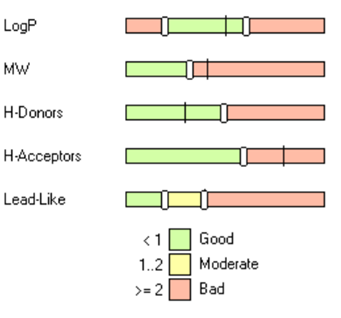
Lead-like (default):
2 violations
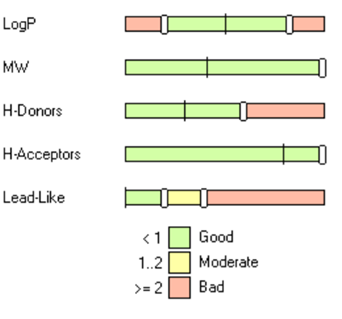
Lead-like (bRo5):
0 violations
Ensuring Accurate pKa Prediction in the bRo5 Space
Among the key physicochemical properties, pKa remains a foundational parameter, directly influencing solubility, permeability, ionization state, and ultimately, pharmacokinetics. Unlike most physicochemical properties, pKa can be viewed as inherently local to each ionizable center, governed by the type of ionizable group and its local chemical environment. This makes pKa more amenable to accurate prediction—even in complex, large, or multifunctional drug candidates—provided the predictive model is trained on diverse and representative data.
ACD/Labs’ PhysChem Suite™ has been developed to meet the evolving needs of researchers working in these non-traditional spaces. Our Classic pKa calculator offers consistent and accurate predictions, even for large and multifunctional molecules by:
Localized Modelling of Ionizable Centers
The algorithm evaluates pKa based on the local structure and context of each ionizable group, rather than treating it as a global molecular property.
Continuously Expanding Training Sets
Recent versions of the ACD/pKa™ Classic algorithm incorporate curated experimental data from novel compound classes, including almost 500 experimental pKa values from over 250 PROTACs and their precursors, significantly improving accuracy across heterobifunctional molecules.
Validating with External Datasets
Performance on independent test sets—including complex PROTACs with ionizable linkers—demonstrates high correlation between predicted and experimental values, reinforcing the model’s reliability for emerging modalities.
Collaborating with Industry Leaders
In partnership with AstraZeneca, ACD/Labs leveraged more than five years of high-quality, experimentally measured pKa data—more than 2,500 values across 1,100 compounds—into its algorithm. This collaboration enabled updates to hundreds of molecular fragments and Hammett equations, and led to the identification of two previously unrecognized dissociating centers. As a result, prediction accuracy improved significantly: 98.7% of pKa values are now predicted within ±1.0 log units; compared to just 72% before refinement. Collaborations such as these enhance predictive accuracy and ensure real-world applicability in modern drug discovery pipelines.
This level of precision enables researchers to confidently explore beyond traditional chemical space, knowing that key ionization behaviors are accurately captured, even in complex molecules. Tools like PhysChem Suite provide strategic insight into ADME challenges, helping chemists prioritize candidates, optimize designs early, and support oral bioavailability in the bRo5 space—all while reducing costly trial-and-error.
Looking Forward
Innovation in drug discovery is advancing rapidly. While modalities like mRNA, CAR-T, and gene editing dominate the headlines, small molecules are simultaneously being redefined. Larger, more flexible compounds with targeted, multifunctional mechanisms are now essential to addressing previously untreatable diseases.
At ACD/Labs, we’re supporting this shift through continuously refined predictive models, collaboration with industry leaders, and a deep commitment to scientific accuracy. Our Percepta software helps research teams worldwide design smarter, more effective drug candidates. As the therapeutic landscape continues to evolve, our tools are already meeting the complexity head-on.
References
- K. R. Hornberger, E. M. V. Araujo. (2023). Physicochemical Property Determinants of Oral Absorption for PROTAC Protein Degraders. J. Med. Chem., 66(12), 8281-8287. doi: 10.1021/acs.jmedchem.3c00740
About the Author
