June 10, 2025
by Mikhail Elyashberg, Leading Researcher, ACD/Labs
Computer Assisted Structure Elucidation of Mauritamide B Using Structure Elucidator Suite
2-aminoimidazole bromopyrrole alkaloids connected by taurine have been isolated previously from marine sponges, particularly from Agelas sp. More than ten compounds of this alkaloid family have been reported so far, including mauritamide B (1) isolated by Hertiani et al. [1]
1
Mauritamide B was isolated from the Indonesian marine sponge Agelas linnaei. The configuration and biological activity of 1 were not described in the original report [1]. Hirozumi and coworkers [2] wanted to confirm the reported structure 1, given that it was determined using an unusual 4JCH HMBC correlation (C-15/ H-19). Despite its structural simplicity, 1 had not yet been synthesized even in its racemic form. To address this gap, authors [2] synthesized racemic 1 to confirm its structure. As a result, it was shown that the true structure of Mauritamide B is instead 2. This was confirmed by total synthesis.

2
In article [3], it was shown on a large number of examples that the use of the ACD/Structure Elucidator program allows for a quick and reliable revision of such structures whose erroneousness was previously determined by synthesis. For structure revision, it was often necessary to synthesize both structures – first the incorrect one, and then the supposedly correct structure. Undoubtedly, a complete synthesis provides convincing confirmation of the structure accuracy. However, the cost of performing total syntheses, especially multi-step ones, is quite high.
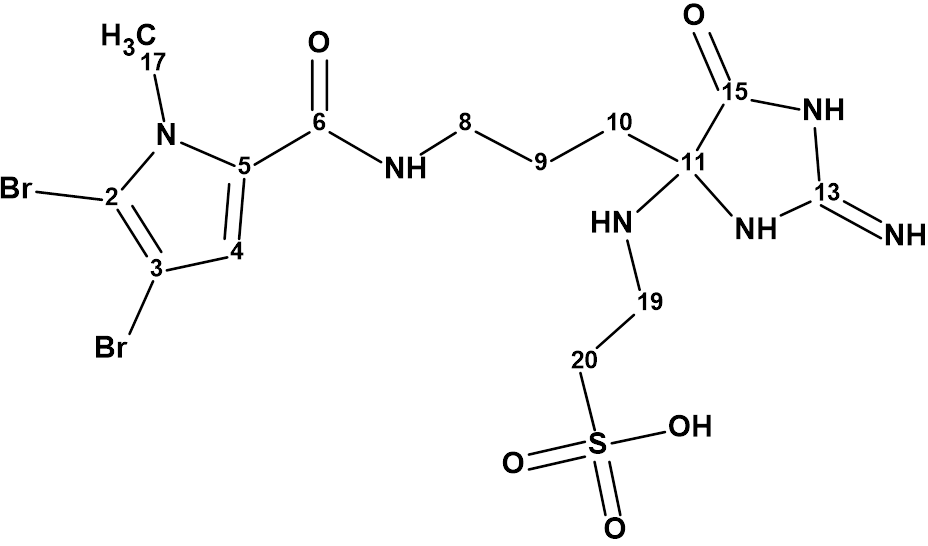
For this reason we attempted to use Structure Elucidator to revise the structure 1 based on the spectroscopic data presented in [1] . The structure of mauritamide B and the results of 13C chemical shift calculations using the methods provided in the program are shown in Figure 1. The average deviations of the calculated chemical shifts from the experimental ones are indicated as follows.
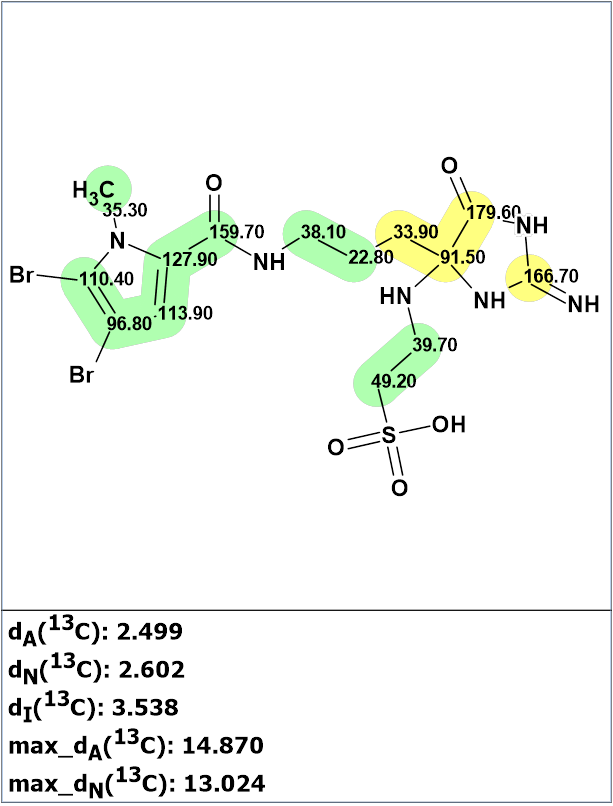
Figure 1. Structure of mauritamide B (1) proposed by Hertiani et al [1] for which 13C chemical shift prediction was carried out using the HOSE code-based method, the neural networks, and the incremental approach. Average deviations of 13C chemical shifts determined by these methods are denoted as dA, dN and dI correspondingly. Each atom is colored to mark a difference between its experimental and calculated 13C chemical shifts. The green color represents a difference between 0 to 3 ppm, yellow was >3 to 15 ppm.
Analysis of the calculation results shows that the values of the average deviations may correspond to the correct structure, but the max_dA ~ 15 ppm value is too large for a correct structure. In addition, the right part of the molecule is colored yellow, which makes structure 1 questionable and requires further investigation.
We see that the structure contains two “green” fragments. We know from experience that this allows us to simplify and speed up the revision of the structure.
The molecular formula C14H20Br2N6O5S and the NMR spectroscopic data of compound 1 shown in Table 1 were entered into the program.
Table 1. NMR spectroscopic data of compound 1.
| Label | δC | δCcalc (HOSE) | CHn | δH | H to C HMBC |
| C 2 | 110.4 | 110.3 | C | ||
| C 3 | 96.8 | 96.75 | C | ||
| C 4 | 113.9 | 113.8 | CH | 6.99 | C 17, C 2, C 5, C 6 |
| C 5 | 127.9 | 127.9 | C | ||
| C 6 | 159.7 | 159.6 | C | ||
| C 8 | 38.1 | 39.13 | CH2 | 3.14 | C 9, C 10, C 6 |
| C 9 | 22.8 | 22.9 | CH2 | 1.27 | C 10, C 8, C 11 |
| C 10 | 33.9 | 36.93 | CH2 | 1.84 | C 9, C 8, C 11, C 15 |
| C 11 | 91.5 | 86.38 | C | ||
| C 13 | 166.7 | 151.83 | C | ||
| C 15 | 179.6 | 171.57 | C | ||
| C 17 | 35.3 | 35.24 | CH3 | 3.86 | C 2, C 5 |
| C 19 | 39.7 | 37.39 | CH2 | 3.6 | C 20, C 15 |
| C 20 | 49.2 | 49.12 | CH2 | 2.74 | C 19 |
The two “green” fragments (highlighted in Figure 2) were entered into the User Fragments library.
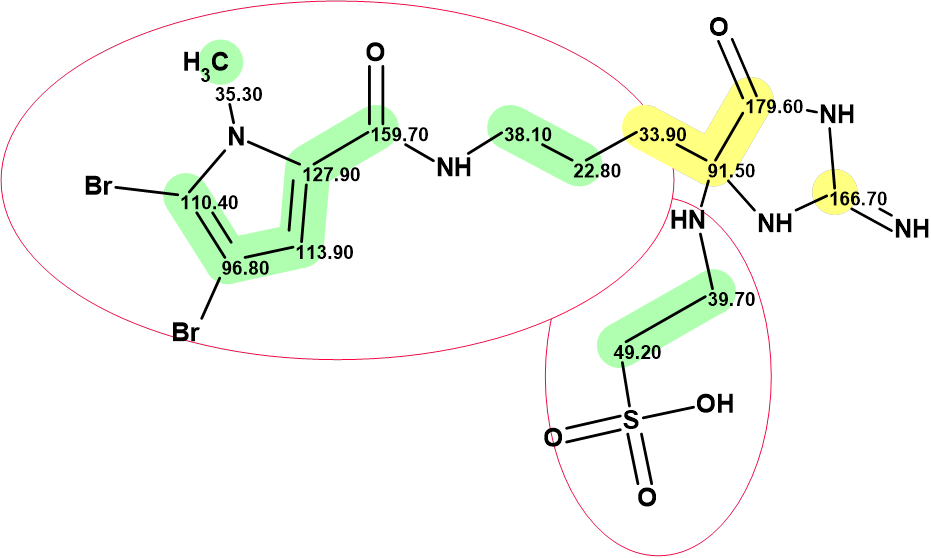
Figure 2. “Green” fragment selection in structure 1.
In order to obtain all conceivable structures during the generation process independent of the constraints imposed by the HMBC, this spectrum was deselected before creating the molecular connectivity diagram (MCD). The MCD is shown in Figure 3.

Figure 3. Molecular connectivity diagram (MCD) of mauritamide B. The MCD contains the two fragments of the proposed mauritamide B structure that were colored green. The hybridizations of carbon atoms are marked by the corresponding colors: sp2 – violet, sp3 – blue, not sp – light blue. The label “ob” is set to carbon atoms for which neighboring with a heteroatom is obligatory (ob). The HMBC connectivities are deselected and not shown. The carbon atom at 91.50 ppm is colored light blue, as this 13C chemical shift value can be because of atoms in both the sp2 or sp3 hybridization states.
The structure generation was launched, accompanied by the prediction of 13C chemical shifts. Result: k = 3.427 → (Structural Filter) → 325 → (Duplicate Removal) → 182, tg = 31 s. The output file was ranked in ascending order of dA deviations. The first 20 structures of the ranked file are shown in Figure 4


Figure 4. The ranked output file. 13C chemical shift prediction was carried out using the HOSE code-based method, the neural networks, and the incremental approach. The average deviations of 13C chemical shifts determined by these methods are denoted as dA, dN and dI correspondingly.
We see that the first structure coincides with the revised structure of mauritamide B which was confirmed by total synthesis [2] . The originally proposed structure 1 was placed in 33rd position by the ranking procedure and not shown in Figure 4. At the same time, all structures are very similar, and the deviation values are small and within the values normally acceptable for a correct structure. Therefore, we calculated the DP4 probabilities for the first 12 structures of the ranked file. The obtained values are shown in Figure 5.


Figure 5. The top 12 structures of the ranked output file. DP4A, DP4N and DP4I are probabilities of structure validity calculated by the program.
Figure 5 shows that the first structure has the highest probability, which confirms the validity of the revision performed in [2]. Additional confirmation of the validity of this structure could be obtained by the calculation of 13C chemical shifts using the DFT approach as shown in [4].
The authors [2] provide the HMBC pattern of compound 2 (Figure 6), which shows that the intensity of the peak C15\H2’ (C15\H19 in structures 1 and 2) is so high that its assignment to the 4JCH correlation is unlikely. If the authors [1] had noticed this they might have established the correct structure of mauritamide B in their first attempt.

Figure 6. The HMBC pattern presented in Supporting Information [2]. All HMBC correlations are of standard length as shown in structure 2.
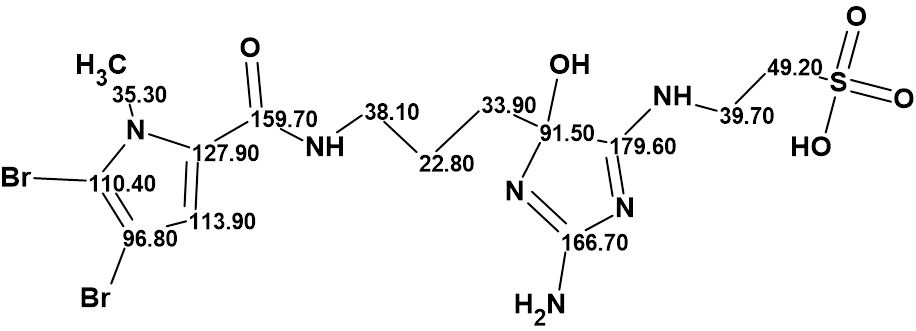
Thus, the revision of the structure of mauritamide B was achieved using Structure Elucidator in half a minute. The validity structure of mauritamide B with assigned 13C chemical shifts is shown below:
References
- Hertiani, T.; Edrada-Ebel, R.; Ortlepp, S.; van Soest, R. W.M.; de Voogd, N. J.; Wray, V.; Hentschel, U.; Kozytska, S.; Muller, W. E.G.; Proksch, P. (2010). Bioorg. Med. Chem., 18(3), 1297−1311.
- Hirozumi, R.; Kudo Y., Cho Y.; Konoki, K.; Yotsu-Yamashita M. (2025). Total Synthesis and Structural Revision of (±)-Mauritamide B. J. Nat. Prod., 88, 806−814.
- Elyashberg, M.; Tyagarajan, S.; Mandal, M.; Buevich, A. V. (2023). Enhancing Efficiency of Natural Product Structure Revision: Leveraging CASE and DFT over Total Synthesis. Molecules, 28(9), 3796.
- Buevich, A. V.; Elyashberg. M. E. (2016). Synergistic combination of CASE algorithms and DFT chemical shift predictions: a powerful approach for structure elucidation, verification and revision. J. Nat. Prod., 79(12), 3105–3116.
About the Author
