March 1, 2016
by Mikhail Elyashberg, Leading Researcher, ACD/Labs
Teotihuacanin
Diterpenoids derived from the Salvia species have shown the capacity to cross membranes and the blood−brain barrier due to their lipophilic properties. These features facilitate interactions with some molecular targets of therapeutic interest. In addition, these compounds have shown cytotoxicity activity against human cancer cells. Diterpenes with unconventional carbon skeletons were isolated from the Salvia
genus.
S. amarissima is an herbaceous plant endemic to and widely distributed throughout Mexico. Recently Bautista et al [1] reinvestigated the chemical composition of S. amarissima. As a result, Teotihuacanin (structure 1), an unusual rearranged clerodane diterpene with a new carbon skeleton containing a spiro-10/6 bicyclic system, was isolated from the leaves and flowers of Salvia amarissima.
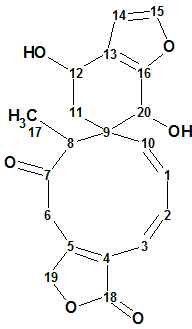
1
The molecular formula (C20H20O6) was deduced from the pseudo-molecular ion peak observed at m/z 357.13406 (HRDARTMS, calculated 357.13381), indicating 11 degrees of unsaturation. The 13C NMR spectrum showed 20 signals, which were resolved through a DEPT experiment as one methyl, three methylenes, nine methines, and seven nonprotonated carbon atoms. In the IR spectrum, bands corresponding to hydroxyl groups (3524 and 3479 cm−1), γ-lactone and ketone carbonyl groups (1744 and 1689 cm−1) were observed.
The authors determined the structure of Teotihuacanin using 1H, 13C, HSQC, HMBC and COSY spectra, and confirmed it via X-ray analysis. The NMR data are collected in Table 1.
Table 1. Spectroscopic NMR data for Teotihuacanin.
| Label | δC | δC Calc | XHn | δH | 1H M | COSY | C HMBC |
| C 1 | 129.5 | 133.38 | CH | 5.82 | u | 6.38, 5.52 | |
| C 2 | 131.5 | 136.8 | CH | 6.38 | u | 5.93, 5.82 | C 4, C 10 |
| C 3 | 118.8 | 123.24 | CH | 5.93 | d | 6.38 | C 18, C 5 |
| C 4 | 123.9 | 128.63 | C | ||||
| C 5 | 157.6 | 149.74 | C | ||||
| C 6 | 41 | 44.6 | CH2 | 3.76 | u | C 7 | |
| C 6 | 41 | 44.6 | CH2 | 2.85 | u | ||
| C 7 | 208.3 | 209.43 | C | ||||
| C 8 | 59.8 | 48.09 | CH | 2.86 | q | 1.24 | |
| C 9 | 47.2 | 46.83 | C | ||||
| C 10 | 133.9 | 136.39 | CH | 5.52 | d | 5.82 | C 9 |
| C 11 | 33.5 | 37.79 | CH2 | 2.35 | d | 4.27 | C 13 |
| C 11 | 33.5 | 37.79 | CH2 | 1.81 | d | ||
| C 12 | 61.9 | 62.21 | CH | 4.27 | u | 2.35 | |
| C 13 | 123.7 | 122.35 | C | ||||
| C 14 | 108.7 | 110.54 | CH | 6.35 | u | 7.26 | |
| C 15 | 142.2 | 142.91 | CH | 7.26 | u | 6.35 | C 13 |
| C 16 | 150.9 | 151.46 | C | ||||
| C 17 | 9.1 | 11.35 | CH3 | 1.24 | d | 2.86 | C 7 |
| C 18 | 172.4 | 172.91 | C | ||||
| C 19 | 73.6 | 73.7 | CH2 | 5.24 | u | C 5 | |
| C 19 | 73.6 | 73.7 | CH2 | 5.24 | u | ||
| C 20 | 65 | 67.71 | CH | 4.43 | s | C 10, C 16, C 13, C 8, C 9, C 11 |
The spectroscopic data were input into ACD/Structure Elucidator Suite from Table 1, and a Molecular Connectivity Diagram (MCD) was created (Figure 1).
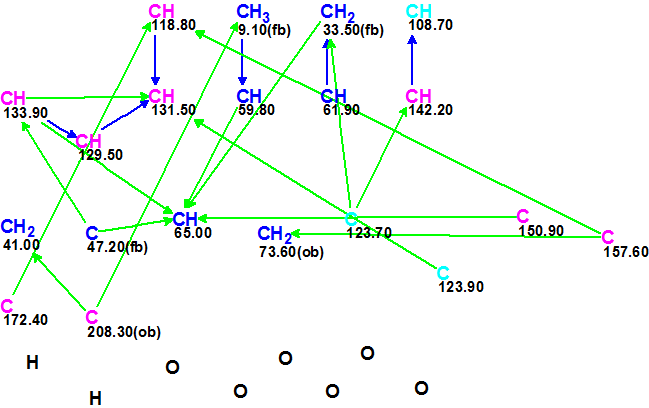
Figure 1. Molecular Connectivity Diagram (MCD) for Teotihuacanin.
MCD overview. Figure 1 shows that three carbon atoms are colored light blue, indicating their hybridization was not determined exactly by the program (sp3 or sp2, but not sp). For some carbon atoms, the possibility of a neighboring heteroatom was automatically set via 13C and 1H chemical shift analysis (ob – obligatory, fb – forbidden). Carbon atoms which are sp3-hybridized are marked in dark blue, but those which are sp2-hybridized are distinguished by violet. Information about signal multiplicities in the 1H spectrum was introduced according to the corresponding data presented in Table 1 (column 1H M). No MCD edits were made.
Checking the MCD for the presence of contradictions showed that all the 2D NMR spectra were internally consistent, and so Strict Structure Generation was initiated, followed by 13C chemical shift prediction and structure filtering (during which the structures for which average deviations d>6 ppm were rejected). The results were: k=46281
→ 49 → 46, tg = 1 m 6 s.
Three top three structures of the output structural file, ranked in increasing order of average 13C chemical shift deviations, are shown in Figure 2.
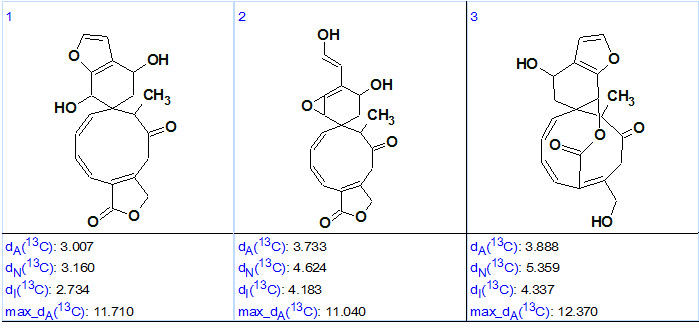
Figure 2. Three top structures of the ranked output structural file for Teotihuacanin.
Figure 2 shows that structure #1 is the best one (based on the lower chemical shift deviations), and it coincides with the structure of Teotihuacanin determined by authors [1] and confirmed by X-ray crystallography. Therefore the correct structure was automatically generated and then selected by the common ranking procedure for ACD/Structure Elucidator Suite.
At the same time, we see that the values of average chemical shift deviations corresponding to three methods of 13C chemical shift prediction (dA – HOSE code based approach, dN – artificial neural nets, dI – incremental approach) are slightly higher than normal for a conclusive structure determination. However, this is likely accounted for by the unusual skeleton of the molecule, as well as by the cis-configuration of both C=C double bonds included in the large cycle of structure #1. Bautista et al [1] established that the C(1) = C(10) bond was a trans-configuration, while C(2) = C(3) was cis-configured. Therefore structure #1 was transformed into its real configuration, and 13C chemical shifts were recalculated (see Figure 3).
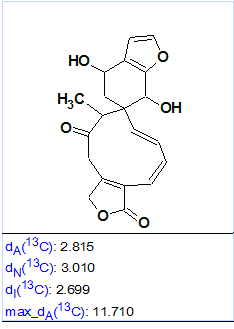
Figure 3. Average chemical shift deviation calculated for structure #1 after bringing its configuration to a real shape.
Figure 3 shows that all of the deviations decreased (compare with those found in Figure 2), as the ACD/CNMR Predictors are sensitive to the C=C double bond configuration. For instance, Figure 4 shows a protocol of 13C chemical shift prediction for the carbon C 133.9 when the HOSE code-based approach was used. We see that the program selects those reference structures for chemical shift calculations that contain double bonds in trans-configuration.
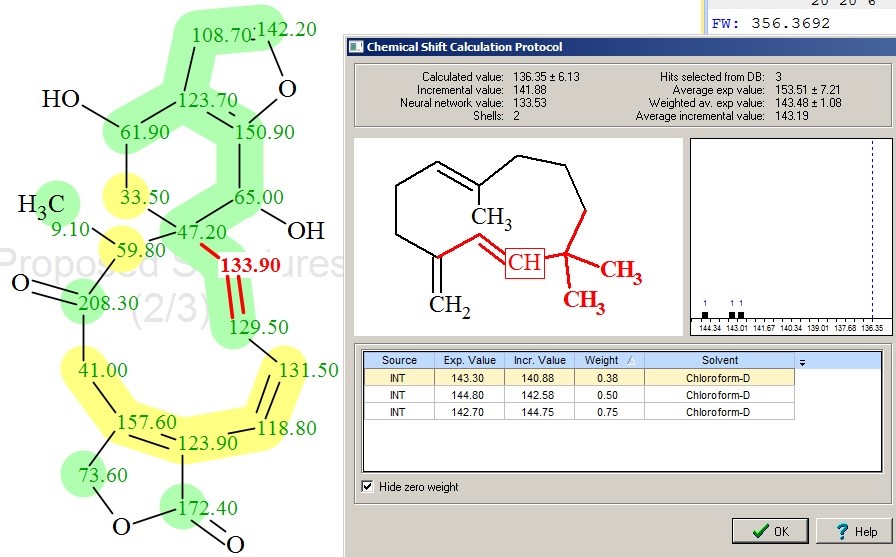
Figure 4. Protocol of 13C chemical shift prediction for carbon C 133.90 for Teotihuacanin. In the structure, the experimental chemical shifts automatically assigned to carbon atoms by the program are shown. The color attributes of the 13C chemical shift prediction accuracy are: ±3 ppm – green, between 3 and 15 ppm – yellow, greater than 15 ppm – red.
Inspection of yellow-colored atoms allowed us to see that the maximum errors in 13C chemical shift prediction correspond to carbons C 59.8 and C 157.6 (compare experimental and predicted (red) chemical shifts in Table 1). The reason for this lies in the wide distribution of chemical shift values in the reference structures used for calculations, which is illustrated by Figures 5 and 6.
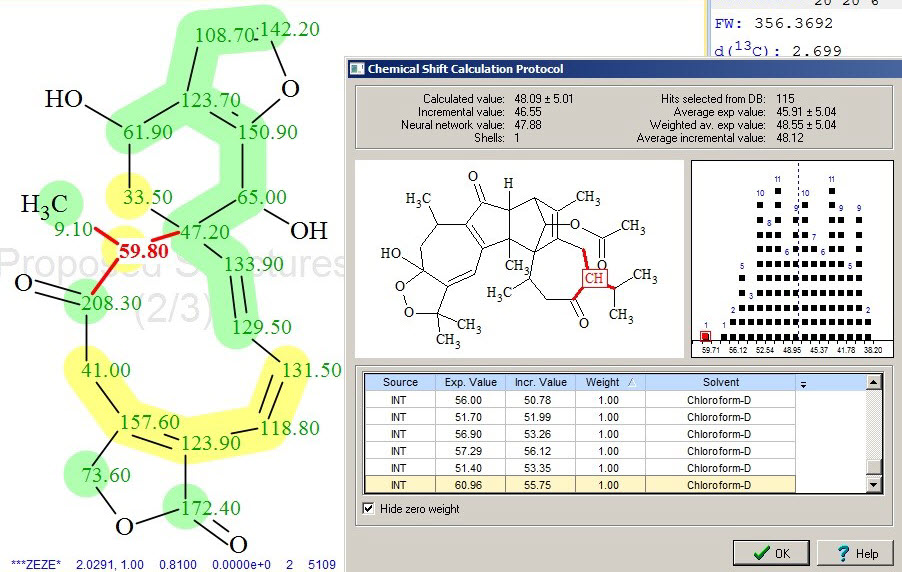
Figure 5. The calculation protocol of 13C chemical shift prediction for C 59.8, showing a single reference structure (one of 115 in total) and the distribution of all chemical shifts used for this calculation.
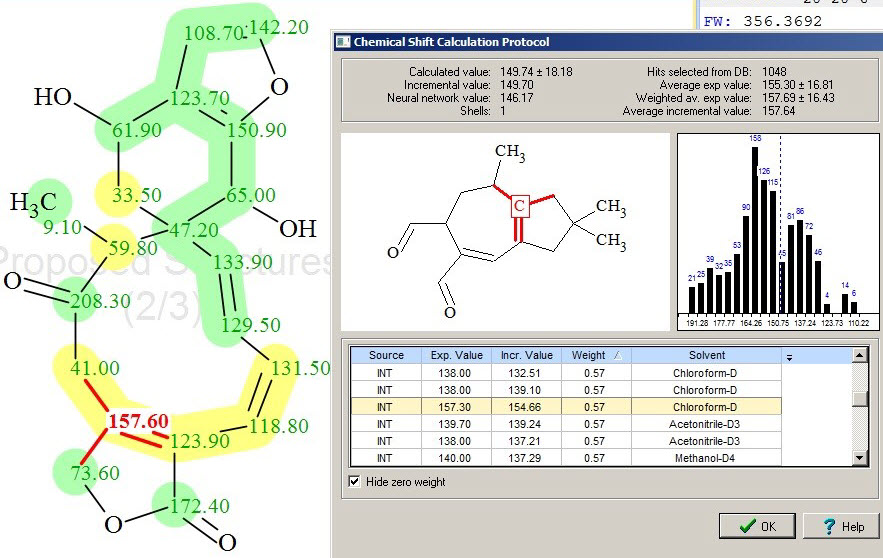
Figure 6. Protocol of 13C chemical shift prediction for C 157.60, showing a single reference structure (one of 1048 in total) and the distribution of chemical shifts used for this calculation.
Therefore this example demonstrates that the structure of a new natural product, possessing unusual skeleton, was elucidated in fully automatic fashion by ACD/Structure Elucidator Suite in one minute.
References
- E. Bautista, M. Fragoso-Serrano, R. A. Toscano, M. del Rosario García-Peña, A. Ortega. (2015). Teotihuacanin, a Diterpene with an Unusual Spiro-10/6 System from Salvia amarissima with Potent Modulatory Activity of Multidrug Resistance in Cancer Cells. Org. Lett., 17: 3280−3282.
About the Author
