August 28, 2023
by Mikhail Elyashberg, Leading Researcher, ACD/Labs
Aspergillamide C
Marine-derived peptides exhibit significant promise for nutraceutical and medicinal applications due to their diverse range of bioactivities, including antimicrobial, antiviral, antitumor, and antioxidative properties. Among these peptides, aspergillamides, which are unique modified tripeptides containing rare dehydrotryptamine moieties, are primarily sourced from marine-derived Aspergillus fungi. Despite displaying moderate cytotoxic effects, these aspergillamides demonstrate structural diversity resulting from geometric isomerization of double bonds and variations in amino acid composition. However, the number of reported natural aspergillamides remains limited to less than ten. To search novel and bioactive secondary metabolites from marine microorganisms, Xiao-Wei and coworkers [1] selected the Aspergillus terreus SCSIO 41008 for chemical investigations. As a result, they isolated and identified a new aspergillamide C (1).
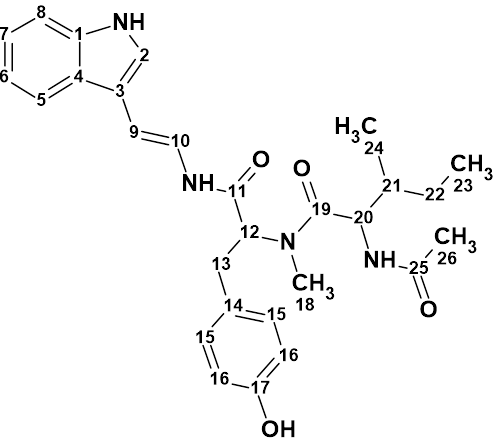
Compound 1 was obtained as a colorless oil. It has a molecular formula of C28H34N4O4 as determined by the quasi-molecular ion peak at m/z 513.2482 [M + Na]+ (Calcd. 513.2478) observed from the HR-ESI-MS data. To elucidate the structure of this compound, 1D NMR, HSQC, HMBC, COSY and NOESY data were used (see Table 1).
Table 1. NMR spectroscopic data used for the structure elucidation of aspergillamide C
| Label | dC | dCcalc (HOSE) | CHn | dH | M (1H) | COSY | H to C HMBC |
| C 1 | 137.6 | 136.7 | C | ||||
| C 2 | 124.3 | 124.6 | CH | 7.64 | s | C 4, C 1 | |
| C 3 | 113.3 | 110.68 | C | ||||
| C 4 | 128.2 | 124.5 | C | ||||
| C 5 | 119.4 | 118.9 | CH | 7.63 | d | 7.08 | C 1 |
| C 6 | 120.5 | 119.4 | CH | 7.08 | u | 7.16, 7.63 | |
| C 7 | 123.1 | 122.25 | CH | 7.16 | u | 7.08, 7.42 | |
| C 8 | 112.4 | 111.9 | CH | 7.42 | d | 7.16 | C 4 |
| C 9 | 105.9 | 106.6 | CH | 6.16 | d | 6.76 | C 2, C 4 |
| C 10 | 118.7 | 119.21 | CH | 6.76 | d | 6.16 | C 11 |
| C 11 | 168.7 | 167.47 | C | ||||
| C 12 | 52.9 | 60.43 | CH | 4.85 | u | 2.83 | C 14, C 11, C 19 |
| C 13 | 38.4 | 33.05 | CH2 | 2.83 | u | 4.85 | C 15 |
| C 13 | 38.4 | 33.05 | CH2 | 2.78 | u | ||
| C 14 | 127.8 | 128.75 | C | ||||
| C 15 | 131.2 | 130.04 | CH | 6.87 | d | 6.64 | C 17 |
| C 16 | 116.5 | 115.01 | CH | 6.64 | d | 6.87 | C 14, C 17 |
| C 17 | 157.4 | 155.92 | C | ||||
| C 18 | 30.7 | 31.13 | CH3 | 2.63 | s | C 19 | |
| C 19 | 175.4 | 172.78 | C | ||||
| C 20 | 62.3 | 53.38 | CH | 4.73 | d | 2.04 | C 18, C 25, C 19 |
| C 21 | 32.7 | 35.77 | CH | 2.04 | u | 0.93, 1.40, 4.73 | |
| C 22 | 25.3 | 24.4 | CH2 | 0.98 | u | ||
| C 22 | 25.3 | 24.4 | CH2 | 1.4 | u | 0.85, 2.04 | |
| C 23 | 10.5 | 11.43 | CH3 | 0.85 | u | 1.4 | |
| C 24 | 15.7 | 14.6 | CH3 | 0.93 | u | 2.04 | |
| C 25 | 172.6 | 170.45 | C | ||||
| C 26 | 22.1 | 22.8 | CH3 | 1.93 | s | C 25 |
These data were entered into ACD/Structure Elucidator (ACD/SE) to challenge the software. A Molecular Connectivity Diagram (MCD) was created by the program and slightly edited manually (see Figure 1).
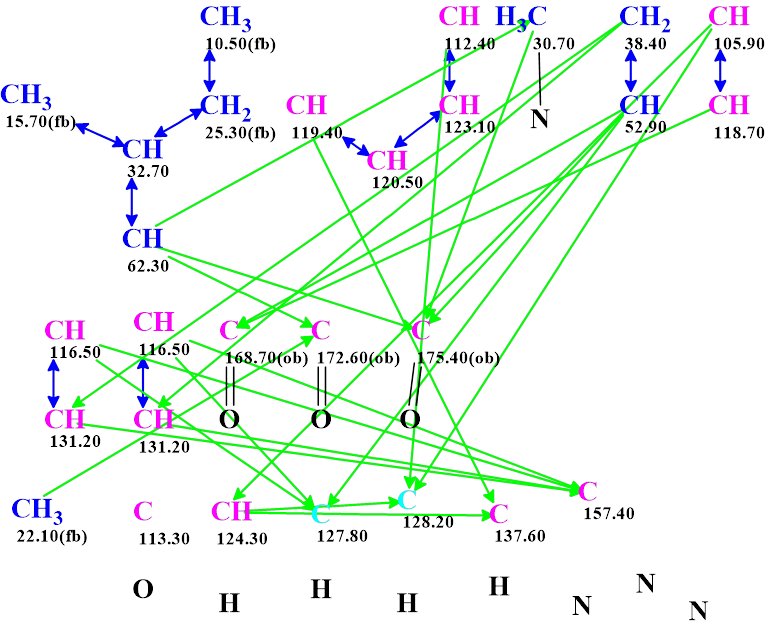
Checking the MCD for the presence of contradictions was completed with the program reporting proper data consistency. Therefore, strict structure generation was initiated, followed by 13C chemical shift prediction and structure filtering. Results: k = 2 162 → (Structure filtering) → 94 → (Duplicate removal) → 71, tg = 11s.
The structures were ranked in increasing order of average deviations of calculated 13C chemical shifts from experimental ones. Interestingly the proposed structure 1 was not generated. The six top ranked structures are shown in Figure 2.
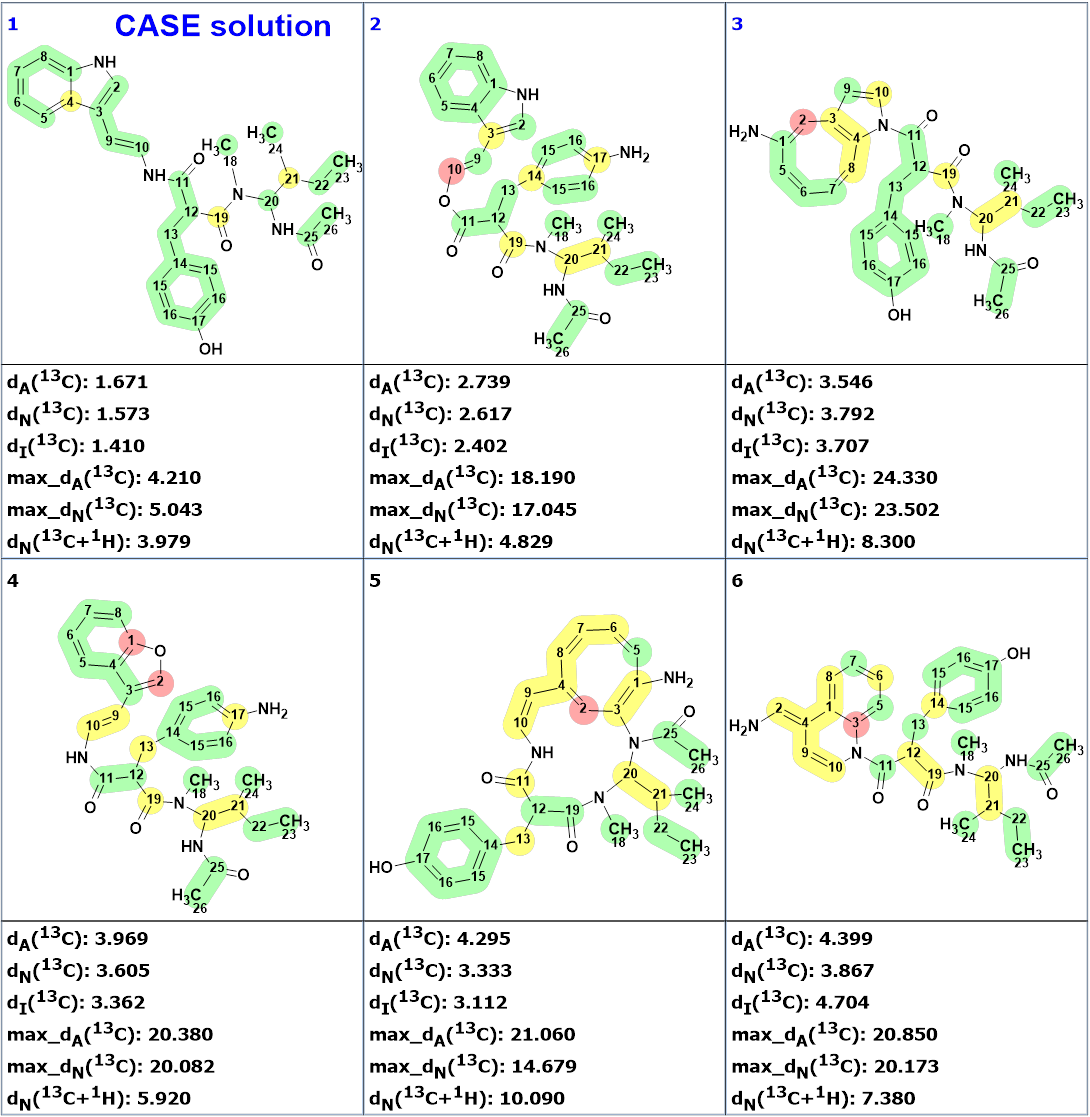
Figure 2 shows that the first ranked structure #1 (CASE solution) differs from compound 1. The difference can be seen clearer in Figure 3.
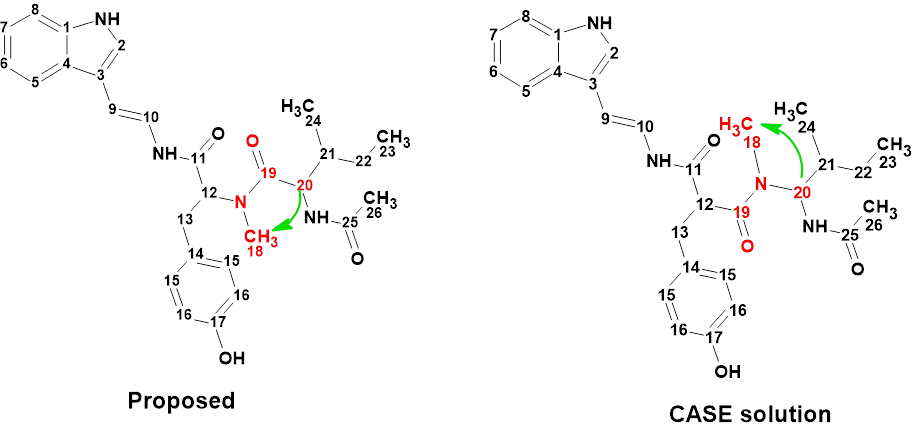
The authors of [1] admitted the presence of a HMBC nonstandard correlation between H-20 and CH3-18, which allowed them to confirm their structural hypothesis. To allow the generation of structure 1, the procedure was repeated with the length of the mentioned correlation set to 2-4 chemical bonds. Results: k = 4276 → (Structure filtering) → 166 → (Duplicate removal) → 121, tg = 19s.
Three top ranked structures are shown in Figure 4.
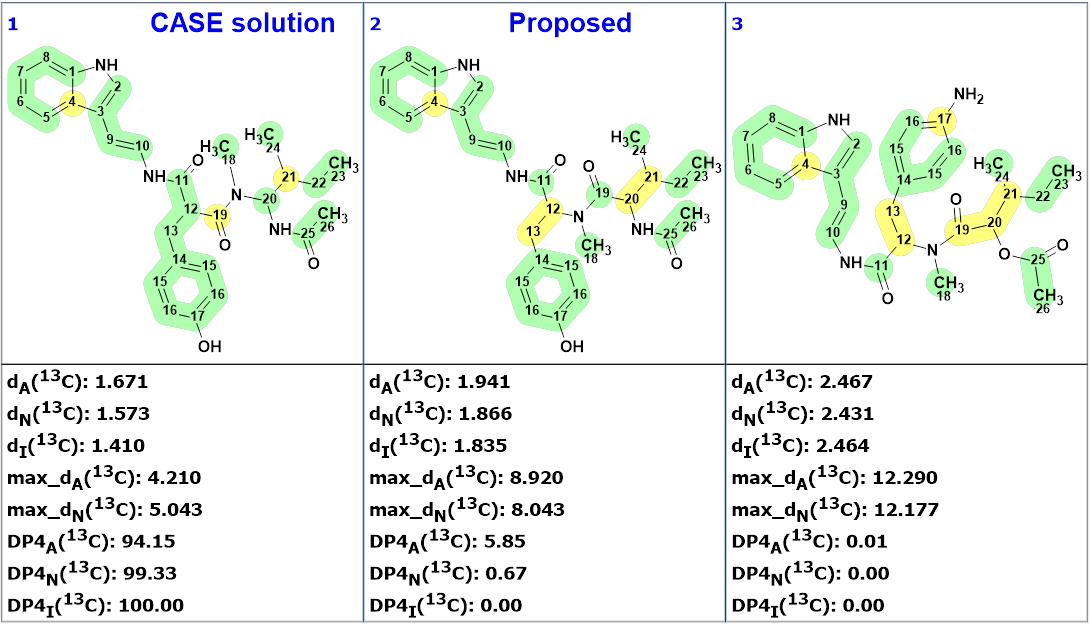
We see that structure 1 was placed in the second position by the ranking procedure and all average and maximum deviations indicate that the CASE solution is the most probable. The validity of this structure was also confirmed by the calculation of DP4 probabilities. According to the methodology suggested and described in works [2-6], the definite determination of the right structure could be done on the basis of conformational analysis and DFT based chemical shift prediction for both structures #1 and #2.
However, the true structure must not only have minimal deviations of the calculated chemical shifts from the experimental values, but also be in agreement with fundamental chemical knowledge. Examples of successful application of this principle to structural revisions were given in a series of papers (e.g., [ 5 ]). Therefore, we discussed the result obtained with Professor Craig Williams, a well-known specialist in the field of chemistry of natural products.
Professor Williams noted that the CASE proposed structure #1 contains an aminal functional group, and these are acid sensitive. In addition, this structure does not make biosynthetic sense, whereas the proposed structure 1 is a functionalized dipeptide. Thus, although the formal features indicate a preference for structure #1 (Figure 4), from a chemical point of view, the structure proposed by the authors of [1] is more likely. Recording 15N correlation experiments (HMBC) could reveal additional information necessary to make the final choice.
This example shows that the knowledge of an organic chemist remains indispensable in the process of structure elucidation and revision.
References
- Xiao-Wei, L.Yun, L. Yong-Jun, Z. Xue-Feng, L. Yong-Hong. (2019). Peptides and polyketides isolated from the marine sponge-derived fungus Aspergillus terreus SCSIO 41008. Chinese Journal of Natural Medicines, 17(2), 149-154.
- V. Buevich, M. E. Elyashberg. (2016). Synergistic combination of CASE algorithms and DFT chemical shift predictions: a powerful approach for structure elucidation, verification and revision. J. Nat. Prod., 79(12), 3105–3116.
- V. Buevich, M. E. Elyashberg. (2018). Towards unbiased and more versatile NMR-based structure elucidation: A powerful combination of CASE algorithms and DFT calculations. Magn. Reson. Chem., 56, 493–504., DOI: 10.1002/mrc.4645
- V. Buevich, M.E. Elyashberg. (2020). Enhancing Computer Assisted Structure Elucidation with DFT analysis of J-couplings. Magn. Reson. Chem., 58(6), 594-606, DOI: 10.1002/mrc.4996
- Elyashberg, I.M. Novitskiy, R. Bates, A.G. Kutateladze, C.M. Williams. (2022). Reassignment of Improbable Natural Products Identified through Chemical Principle Screening. Eur. J. Org. Chem., e202200572. https://doi.org/10.1002/ejoc.202200572
- Elyashberg, S. Tyagarajan, M. Mandal, A.V. Buevich. (2023). Enhancing Efficiency of Natural Product Structure Revision: Leveraging CASE and DFT over Total Synthesis. Molecules, 28(9), 3796.
About the Author

Sometimes a circle inside the hexagon is used instead of the conjugated double bonds to represent the six pi electrons with alternating single and double bonds. If the circle loses one electron to become a radical, can we still use the ACD Lab software to draw the circle with a dot at the centre to represent alternating radical points in the circle?
I believe you can do that using the following steps: first draw the structure using the single and double bound representation. Next, select the section of the molecule where you would like to show delocalization, and click the “Delocalize Curve” button, which can be found in the top bar. Next, using the radical tool that can be found in the bottom left.
Hope you used the button “Solid Delocalization Curve” mentioned by Jesse successfully.