March 1, 2014
by Mikhail Elyashberg, Leading Researcher, ACD/Labs
Barmumycin
Natural products from terrestrial plants and microorganisms have long been a traditional source of drugs; however, over the past few years, marine organisms have garnered ever-increasing attention as a rich bank of new bioactive compounds. Marine actinomycetes have also proven to be an important source of biologically active compounds.
Among the marine actinomycetes those of the genus Streptomyces have clearly shown the most pharmacological potential; however, in many bioactive cultures they have yielded only compounds that are already known. During ongoing research efforts to explore the biosynthetic potential of rare marine microorganisms, Lorente and co-workers [1] isolated two known compounds, pretomaymycin and oxotomaymycin, plus the previously unknown compound barmumycin from the culture broth of the marine actinomycete Streptomyces sp. BOSC-022A, isolated from a tunicate collected off the Scottish coast. Barmumycin and its diacetate show antitumor activity at micromolar concentrations. The authors reported the isolation, total synthesis, and structure elucidation of Barmumycin [1].
The molecular formula of Barmumycin was determined to be C15H19NO4 by HRMS MALDI-TOF; it gave an (M+H)+ ion at m/z 278.13840 (calculated m/z 278.13869 for C15H20NO4). The IR absorption band at 3288 cm-1 gives a hint to the presence of OH/NH groups, while the bands at 1600 and 1585 cm-1 readily indicate the presence of a benzene ring. Reaction of barmumycin with acetic anhydride and pyridine gave a diacetyl derivative, which was confirmed by MS, indicating the presence of two OH and/or NH protons. 1H NMR shows three groups of protons. The first group is in the aromatic region and contains three protons at 6.90 (d), 7.03 (d), and 7.08 (s) ppm; the chemical shifts indicated a 1,2,4-substituted benzene ring. The second group corresponds to a single vinylic proton at 5.34 ppm (m). The third region comprises an upfield CH shift at 4.67 ppm (m); two methyl groups, seen at 3.90 ppm (s) and 1.62 ppm (d); and the signals of three CH2 at 4.07 (bt), 4.18 (bd), 3.75 (m), 2.29 (m) and 2.71 (m) ppm. On the basis of these data, the MS findings, and further data from one-dimensional and two-dimensional (COSY, HMBC, and NOESY) 1H and 13C NMR experiments, the authors [1] initially proposed that the structure of the isolated natural product was that of benzomacrolactone (1), derived from 5-methoxy-2-aminobenzoic acid with an exocyclic (E)-ethylidene and one alcohol function.
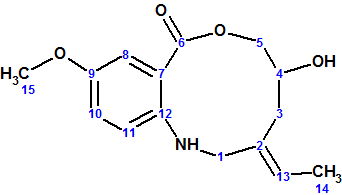
1
To confirm this assignment, compound 1 was synthesized following two different strategies starting from an o-aminobenzoic ester: one (strategy A) based on reductive amination, and one (strategy B) based on N-alkylation, which was shorter and higher yielding.
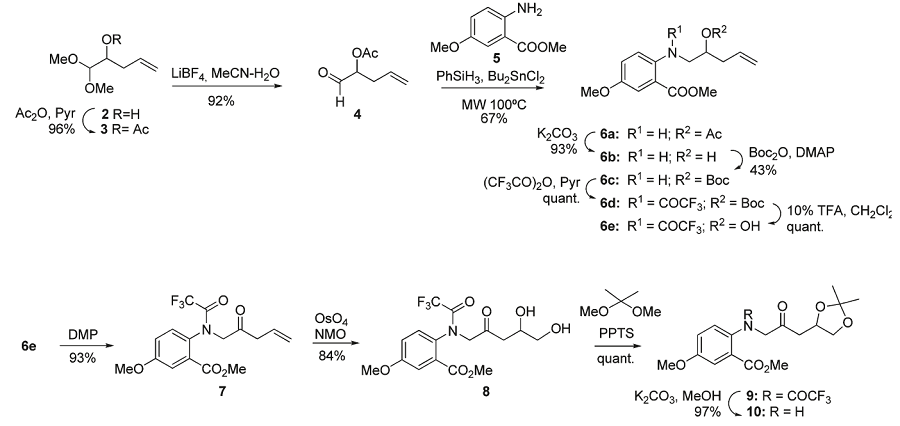
Strategy A
Click to view larger
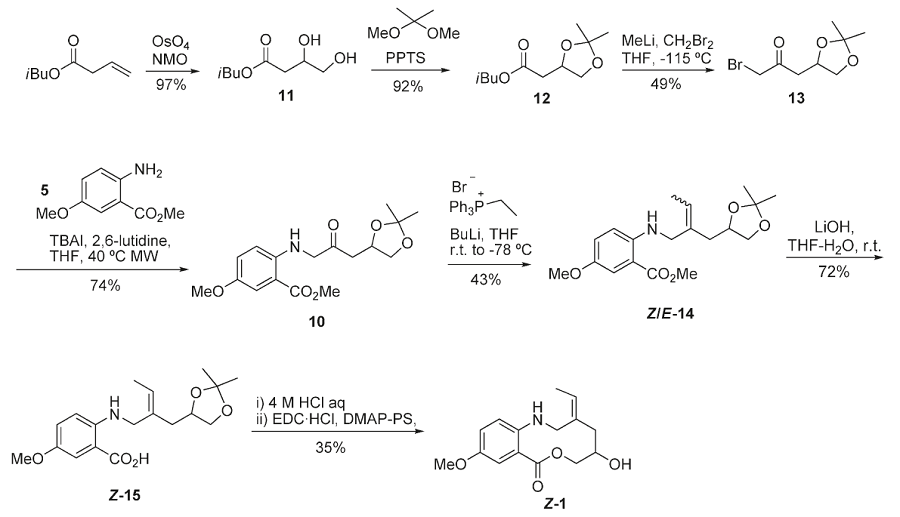
Strategy B
Click to view larger
However, comparison of the NMR spectra for 1 with those for isolated Barmumycin showed dramatic differences. The structure of Barmumycin was reassessed, and most probable option conceived was compound 2 (Z/E-16 in the original article [1]).
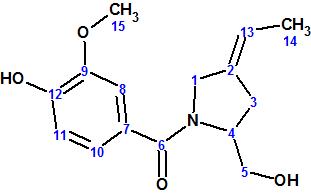
2
Compound 2 was subsequently prepared (in five steps and 18% overall yield) for comparison with the natural compound (see Strategy C below).
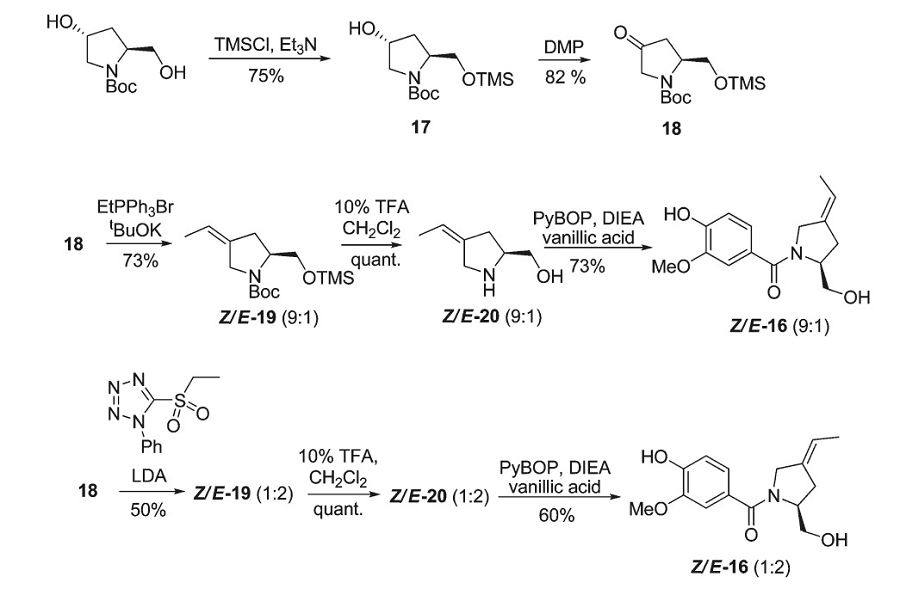
Strategy C
Click to view larger
The spectroscopic data for 2 fully coincided with that for Barmumycin, thereby confirming that the two structures are equivalent.
We investigated how the structure elucidation of Barmumycin could be carried out if ACD/Structure Elucidator Suite was used from the very beginning. The spectroscopic data (13C, 1H , HSQC and HMBC) available from the article [1] are collected in Table 1:
Table 1. Barmumycin. Spectroscopic data.
| Label | δC | CHn | δH | M | C HMBC |
| C 1 | 55.1 | CH2 | 4.11 | u | |
| C 2 | 134.7 | C | |||
| C 3 | 30.1 | CH2 | 2.72 | u | |
| C 3 | 30.1 | CH2 | 2.27 | u | |
| C 4 | 60.6 | CH | 4.67 | u | |
| C 5 | 67.1 | CH2 | 3.75 | u | |
| C 6 | 172.2 | C | |||
| C 7 | 128.3 | C | |||
| C 8 | 110.6 | CH | 7.09 | s | C 6, C 12, C 10, C 9 |
| C 9 | 146.7 | C | |||
| C 10 | 121 | CH | 7.04 | d | C 8, C 12 |
| C 11 | 114.1 | CH | 6.91 | d | C 9, C 7 |
| C 12 | 147.8 | C | |||
| C 13 | 117.6 | CH | 5.34 | u | |
| C 14 | 14.5 | CH3 | 1.62 | u | C 2, C 13 |
| C 15 | 56.3 | CH3 | 3.9 | u | C 9 |
The edited Molecular Connectivity Diagram (MCD) created from the spectroscopic data from Table is shown in Figure 1:
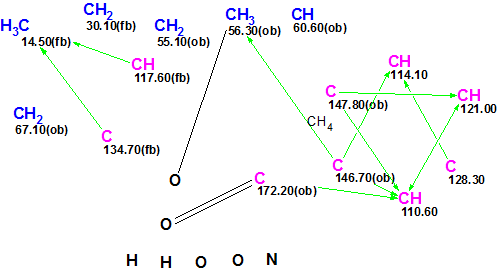
Figure 1. Edited Molecular Connectivity Diagram.
MCD Overview. The number of HMBC connectivities is small. In addition, four carbon atoms have no connectivities at all. So we can expect that the structure generation will be quite time consuming. Therefore some atom properties which follow from spectrum-structure empirical correlations [2] were manually added to MCD. Here the type of atom hybridization is marked in its own specific color: sp3 – blue, sp2 – violet. Labels “ob” and “fb” show if the neighborhood with heteroatoms are obligatory (ob) or forbidden (fb). The evident carbonyl double bond and methoxy group were also manually added.
Structure generation accompanied with 13C chemical shift prediction and spectral filtering (structures for which the deviation value was d>5 ppm were rejected) gave the following results: k= 73373→1089→ 778, tg= 3 m 17 s.
Eight top structures of the output file ranked in ascending order of deviation value are shown in Figure 2, where accuracy of chemical shift prediction for carbon atoms is marked by corresponding colors: d<3 ppm – green, 3<d>15 ppm – yellow.
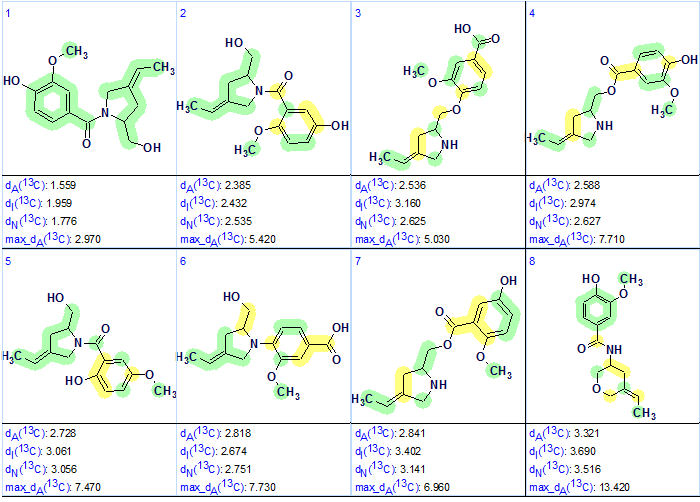
Figure 2. Eight top structures of the ranked output file.
Figure 2 shows that the first ranked structure coincides with the revised structure of Barmumycin 2. All atoms of the structure are marked by green circles, which underlines the best structure priority. The original structure 1 was only ranked as 45th as shown in Figure 3:
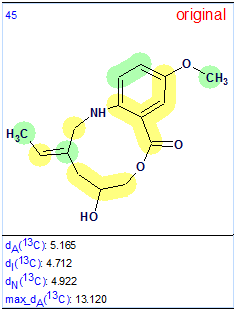
Figure 3. The original structure (1) along with its average and maximum deviations. In contrast to the revised structure, majority of carbon atoms of the original structure are marked with yellow circles.
The original and revised structures supplied with 13C chemical shift assignments automatically performed by the program are shown for comparison in Figure 4:
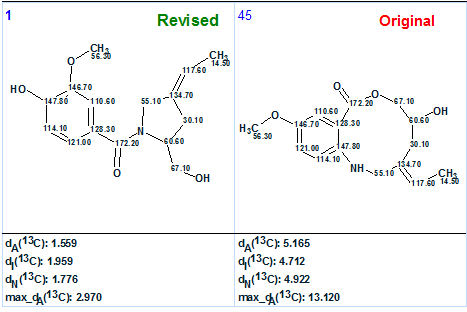
Figure 4. Original and revised structures. Comparison of 13C chemical shift assignments and average deviations calculated for both structures.
In the end of the articles authors [1] conclude that “this work is a new example of the importance of total synthesis for structural characterization and confirmation of natural products.” We do not contest this statement. We should like only to add some comments reflecting our point of view on methodology of structure revision. This example shows that if ACD/Structure Elucidator Suite was employed for elucidation of the Barmumycin structure from the very beginning, the right structure would be unambiguously found in several minutes. The wrong original structure which occupied 45th position in the ranked output file would be not involved in the game at all, and consequently it would be not necessary to carryout its total synthesis. And finally, if at least the empiric 13C NMR chemical shift prediction were performed for the originally supposed structure, the erroneous structural hypothesis would be immediately rejected.
References
- Isolation, Structural Assignment, and Total Synthesis of Barmumycin, A. Lorente, D. Pla, L. M. Cañedo, F. Albericio, M. Alvarez., J. Org. Chem. 2010, 75:8508–8515.
- Pretsch, E.; Clerc, T. E.; Seibl, J.; Simon, W. Tables of Spectral Data for Structure Determination of Organic Compounds; Springer-Verlag: Berlin, 1989.
About the Author
