March 1, 2018
by Mikhail Elyashberg, Leading Researcher, ACD/Labs
Coniothyrione
Coniothyrione is a natural product with a molecular formula of C14H9ClO6. The ratio of the number of heavy atoms to hydrogen is 2.3 indicating an especially challenging structure elucidation [1]. The original structure of coniothyrione, 1, was proposed based on the analysis of the 1H and 13C NMR data, and HMBC correlations [2] summarized in Table 1.
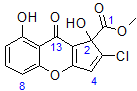
1
Table 1. NMR spectroscopic data for coniothyrione (1)
| Label | δC | δC calc[a] | XHn | δH | J in Hz | HMBC (H to C) |
| C 1 | 168.7 | 172.74 | C | |||
| C 2 | 79.8 | 78.01 | C | |||
| C 3 | 127.2 | 132.49 | C | |||
| C 4 | 143.1 | 129.25 | CH | 7.2 | s | C 2, C 5, C 14 |
| C 5 | 164.5 | 154.7 | C | |||
| C 7 | 155.7 | 156.53 | C | |||
| C 8 | 108.1 | 108.9 | CH | 7.22 | dd,8.5, 1.0 |
C 10, C 12 |
| C 9 | 135.8 | 137.35 | CH | 7.7 | t, 8.5 | C 7, C 11 |
| C 10 | 112.7 | 112.14 | CH | 6.9 | dd,8.5, 1.0 | C 8, C 12 |
| C 11 | 160.6 | 160.56 | C | |||
| C 12 | 110.7 | 109.78 | C | |||
| C 13 | 176 | 175.24 | C | |||
| C 14 | 120.8 | 114.22 | C | |||
| C 15 | 52.9 | 51.97 | CH3 | 3.64 | s | C 1 |
| O 1 | OH | 12.4 | s | C 10, C 11, C 12 |
The lack of a three-bond HMBC correlation from the olefinic proton, H4 (δH 7.20 ppm), to the carbonyl group (C1) in the HMBC spectrum was interpreted by the authors [2] as evidence that the olefinic proton was connected to the carbon C4 and the chlorine was attached to carbon C3. However, as is generally known, the absence of an HMBC correlation between a given proton and a carbon does not necessarily mean that the number of bonds between them is larger than three [3]. There are several cases where one expects to see an HMBC peak for a given pair of nuclei and, for some reasons, this does not appear.
Kong and co-workers [4] noted that the lack of the aforementioned three-bond HMBC correlation could be due to an unfavorable dihedral angle between the olefinic proton and the methoxycarbonyl group (C1). The dihedral angle was found to be approximately 70°ree; by MM2 force field calculation. The change of the olefinic proton position from C4 to C3 in structure 2 did not violate any HMBC correlations described in the article [2]. The revision of the structure of coniothyrione to 2 by Kong and co-workers [4] was based on empirical chemical shift arguments and a speculative biosynthetic pathway without any experimental data. Subsequently, Martin and co-workers [5] undertook a more rigorous experimental and DFT-based theoretical investigation of coniothyrione. The structure of coniothyrione as 2 was confirmed with much stronger proof, which included 1,1-ADEQUATE experiments and DFT analysis of JCC, JHC couplings, and carbon chemical shifts [5].
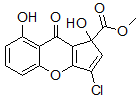
2
The NMR spectroscopic data for coniothyrione presented in Table 1 were analyzed by ACD/Structure Elucidator with a marginally edited Molecular Connectivity Diagram (MCD) as shown in Figure 1. The following edits were made to MCD: four carbons with 13C chemical shifts in the range between 160.6 and 176.6 ppm were labeled as ob, which indicated that at least one heteroatom must be a neighbor of the corresponding carbon atom. Note, that the carbon atom at 127.2 ppm did not have any connectivities to other atoms, so a great number of generated structures was expected.
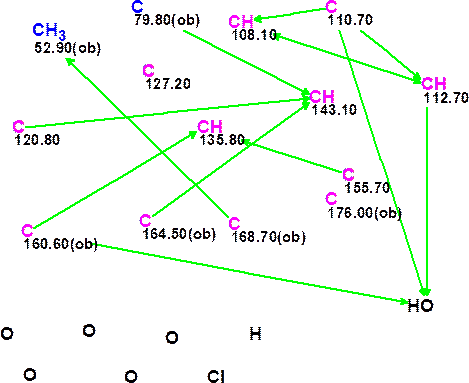
Figure 1. Molecular Connectivity Diagram for coniothyrione.
There were no contradictions detected in HMBC data, and Strict Structure Generation algorithm gave the following results: k = 157,803 → 36,590 → 14,986, tg = 1 min, where k is the number of generated structures (157,803) from which 36,590 passed spectral and structural filtering [1], while 14,986 structures were finally saved after removing duplicates, and tg is the time used for structure generation. Thus, there were approximately 15,000 structures that satisfied both the constraints displayed in MCD and system knowledge. It is worth noting that when the ACD/Structure Elucidator solutions to more than 400 problems were examined (solutions to about 100 new problems were described in [1]), it was found that the correct structure was usually ranked as the first or at least among the top three candidates [1,6]. Only once was the correct structure as low as fifth. These previous results justified the selection of the six top-ranked structures for further analysis (Figures 2).

Figure 2. Six top-ranked structures from CASE study of coniothyrione.
CASE analysis of coniothyrione showed that the revised structure 2, was the second-ranked structure #2 shown in Figure 2; average and maximum deviations of the top six structures were too close to make a final decision regarding the correct structure of coniothyrione based on those criteria. Note, that each predicted structure in Figure 2 from #3 to #6 is expected to have a relatively large three-bond J-coupling between the isolated vinyl proton (H4) and one of the low field carbonyl/carboxyl atoms. This argument is justified by the fact that these atoms and those separating them are coplanar. Since this correlation was not experimentally detected (Figure 1) these structures could be discarded from further analysis. Interestingly, the original structure, 1, was placed in the 18th (!) position by the ranking procedure (see Figure 3). Its average deviations and max_dA=25.55 ppm allowed the structure to be confidently rejected from consideration.

Figure 3. The twenty top-ranked structures of the output file.
It is noteworthy, that the top two structures shown in Figure 2, #1 and #2, and displayed separately in Figure 4 differ only by the orientation of the two well characterized but nearly independent subunits.
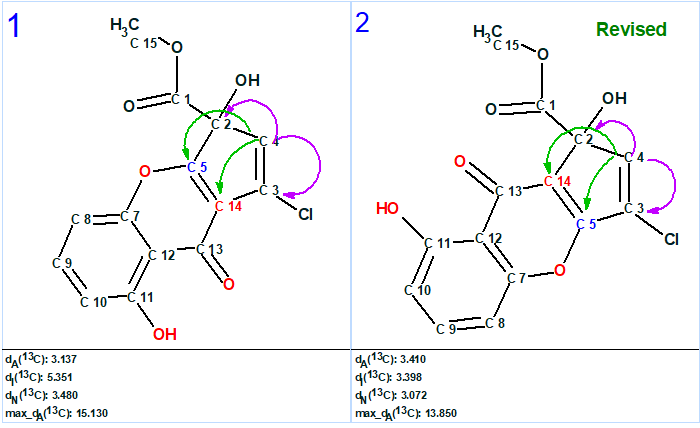
Figure 4. The two top structures of the output file. Green arrows denote HMBC connectivities, violet arrows denote 1,1-ADEQUATE connectivities.
None of the structures had any contradiction with the MCD and both fully satisfied all 2D NMR correlations data used in the structure revision study [5] and, therefore, should be considered as viable candidates. An attempt to differentiate structures #1 and #2 from the other four structures from CASE analysis with the DFT approach was undertaken. The summary of DFT calculations for the top six highest ranked by CASE structures is shown in Table 2.
| Exper. | Structure #1 | Structure #2 (2) | Structure #3 | Structure #4 | Structure #5 | Structure #6 | |
| Labels | δC | δCcalc | δCcalc | δCcalc | δCcalc | δCcalc | δCcalc |
| 1 | 168.7 | 171.3 | 172.0 | 165.9 | 165.6 | 164.0 | 160.4 |
| 2 | 79.8 | 81.6 | 79.6 | 69.6 | 85.1 | 84.4 | 102.9 |
| 3 | 127.2 | 142.7 | 135.6 | 146.1 | 147.1 | 140.9 | 156.3 |
| 4 | 143.1 | 125.4 | 144.9 | 134.1 | 135.8 | 136.1 | 125.7 |
| 5 | 164.5 | 171.0 | 163.8 | 160.8 | 155.7 | 163.8 | 164.1 |
| 7 | 155.7 | 154.6 | 154.7 | 158.8 | 160.3 | 159.6 | 154.7 |
| 8 | 108.1 | 105.3 | 105.5 | 105.3 | 101.5 | 103.8 | 105.7 |
| 9 | 135.8 | 134.8 | 134.3 | 125.3 | 126.4 | 126.7 | 134.1 |
| 10 | 112.7 | 111.9 | 112.0 | 106.5 | 108.9 | 109.1 | 111.8 |
| 11 | 160.6 | 160.5 | 160.5 | 145.3 | 150.2 | 148.9 | 160.8 |
| 12 | 110.7 | 109.8 | 110.3 | 110.2 | 108.8 | 108.6 | 110.1 |
| 13 | 176.0 | 173.9 | 172.9 | 159.1 | 173.1 | 180.5 | 172.3 |
| 14 | 120.8 | 117.2 | 120.6 | 121.1 | 124.7 | 122.7 | 117.3 |
| 15 | 52.9 | 54.1 | 53.7 | 53.8 | 53.2 | 51.7 | 52.0 |
| RMSD | – | 6.75 | 2.76 | 9.46 | 7.86 | 6.40 | 11.29 |
| max_d | – | 17.71 | 8.39 | 18.94 | 19.87 | 13.67 | 29.07 |
RMSD and max_d for structure #2 were significantly lower than those of other candidate structures (Table 2), which confirmed the revised structure 2 of coniothyrione. Even though the structure 2 can be credibly assigned based on DFT predictions of carbon chemical shifts, it is worth noting that, the RMSD and max_d of structure #2 (2) can be further improved by inclusion of relativistic corrections for carbon C3 or by applying the WC04 functional, as previously described [5]. It has also been suggested [7], the chemical shift of the C3 carbon can be excluded from the analysis, and then the RMSD and, in particular, max_d parameter for structure #2 (2) would drop down to a more acceptable values of 1.67 ppm and 3.33 ppm, respectively.
Further differentiation of structures #1 and #2 (2) of coniothyrione could be also done by the heteronuclear J-coupling analysis. Thus, the DFT-calculated J(H3,C5) and J(H3,C14) coupling constants for structure #2 (2) (12.7 and 4.1 Hz, respectively) were in nearly perfect agreement with experimental values of 12.1 and 4.4 Hz [5], whereas the predicted J-couplings for structure #1 of 8.6 and 8.0 Hz were inconsistent with experimental values.
In summary, application of the CASE algorithm in the original study of coniothyrione would have prevented the publication of the erroneous structure while suggesting a very plausible alternative structure that has not been tested before, but should have been due to its full conformity with experimental data. Only the subsequent application of DFT calculations has confirmed that the revised structure 2 was indeed the correct structure.
References
- Elyashberg, M. E.; Williams, A. J. Computer-based Structure Elucidation from Spectral Data. The Art of Solving Problems; Springer, Heidelberg, 2015.
- Ondeyka, J. G.; Zink, D.; Basilio, A.; Vicente, F.; Bills, G.; Diez, M. T.; Motyl, M.; Dezeny, G.; Byrne, K.; Singh, S. B. J. Nat. Prod. 2007, 70, 668- 670.
- Elyashberg, M. E.; Williams, A. J.; Blinov K. A. In Modern NMR Approaches to the Structure Elucidation of Natural Products, Instrumentation and Software. Williams, A.; Martin, G.; Rovnyak, D. Eds., RSC, Cambridge, 2015, Vol. 1, Chapter 9, pp 187-242.
- Kong, F.; Zhu, T.; Pan, W.; Tsao, R.; Pagano, T. G.; Nguyen, B.; Marquez, B. Magn. Reson. Chem. 2012, 50, 829-833.
- Martin, G. E.; Buevich, A. V.; Reibarkh, M.; Singh, S. B.; Ondeyka, J. G.; Williamson, R. T. Magn. Reson. Chem. 2013, 51, 383-390.
- Elyashberg, M. E.; Blinov, K. A. ;Williams, A. J.; Molodtsov, S. G. ; Martin ,G. E. J. Chem. Inform. Model. 2006, 46, 1643-1656
- Lodewyk, M. W.; Siebert, M. R.; Tantillo, D. J. Chem. Rev. 2012, 112, 1839-1862.
About the Author
