September 1, 2018
by Mikhail Elyashberg, Leading Researcher, ACD/Labs
Cycloshermilamine D
The pyridoacridines appear to be the largest group among marine alkaloids, mainly being isolated from tunicates and sponges. A large variety of these compounds has been isolated from the genera Cystodytes. Investigation of Cystodytes resulted in the isolation of five pyridoacridines, including shermilamines D and E. The structure of another compound, cycloshermilamine D (1), also isolated from the same tunicate in minute amounts (0.4% of the crude extract) was determined by Koren-Goldshlager et al [1].
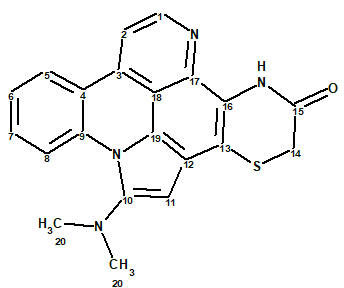
1
Cycloshermilamine D (1) was obtained from the CHCl3-MeOH (1:1) extract, after chromatography on Sephadex LH-20 and Silica gel columns, as a yellow amorphous powder.
The molecular formula of 1 was determined by HR-EI-MS (m/z 372.1045, 100%, M+, Dmmu = -0.1) and NMR data to be C21H16N4OS, with 16 units of unsaturation, indicating an aromatic structure. An IR absorption at 1682 cm-1, together with the 163.4 ppm 13C signal, suggested an amide as in the shermilamines. The ratio of skeletal atoms to hydrogens is 1.7, showing a severe deficiency of hydrogen atoms. Therefore structure elucidation of this molecule is expected to be challenging.
The molecular formula and 1D and 2D NMR data (Table 1) presented in the article [1] were entered into ACD/Structure Elucidator.
Table 1. Spectroscopic NMR data.
| Label | δC | δC calc (HOSE) |
CHn | δH | M,J(1H) | COSY | H to C HMBC |
| C 1 | 143.6 | 146.37 | CH | 8.83 | d, 5.2 | 7.96 | C 2, C 3, C 17 |
| C 2 | 109.2 | 108.99 | CH | 7.96 | d, 5.2 | 8.83 | C 18, C 4 |
| C 3 | 131.5 | 131.5 | C | ||||
| C 4 | 120.5 | 120.61 | C | ||||
| C 5 | 125.7 | 124.19 | CH | 8.47 | d,8.0 | 7.55 | C 3, C 7, C 9 |
| C 6 | 124.6 | 124.8 | CH | 7.55 | t,7.5 | 7.80, 8.47 |
C 8, C 4, C 9 |
| C 7 | 132.5 | 130.8 | CH | 7.8 | t,7.5 | 7.55, 9.13 |
C 5, C 9 |
| C 8 | 118.5 | 118.4 | CH | 9.13 | d,8.0 | 7.8 | C 4, C 6, C 9 |
| C 9 | 136.5 | 135.68 | C | ||||
| C 10 | 150.1 | 146.09 | C | ||||
| C 11 | 94.3 | 86.02 | CH | 6.78 | s | C 19, C 10 |
|
| C 12 | 115 | 122.67 | C | ||||
| C 13 | 113.8 | 113.83 | C | ||||
| C 14 | 29.6 | 28.94 | CH2 | 3.64 | s | C 13, C 15 |
|
| C 15 | 163.4 | 162.86 | C | ||||
| C 16 | 120.3 | 122.69 | C | ||||
| C 17 | 137.3 | 133.24 | C | ||||
| C 18 | 112.6 | 107.5 | C | ||||
| C 19 | 118.8 | 123.44 | C | ||||
| C 20 | 45.2 | 41.89 | CH3 | 2.96 | s | C 10 |
The Molecular Connectivity Diagram (MCD) produced automatically by the program is shown in Figure 1.
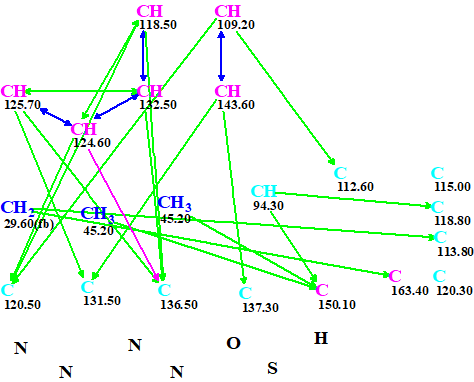
Figure 1. An automatically created molecular connectivity diagram. A violet arrow marks a HMBC connectivity for which the distance between intervening nuclei may vary from one to four bonds.
MCD overview. The MCD contains 10 carbons colored in light blue color, which means that hybridization of these atoms is either sp3 or sp2 (not sp). Therefore ~ 1000 (210) combinations of carbon atom hybridizations should be considered by the program. In addition, two light blue atoms (115.00 and 120.3 ppm) are not involved in the connectivity net. There are also four nitrogen and one sulfur atom that are in the same sense “free”, meaning there are no known correlations of them to any other atoms. All mentioned observations lead to the conclusion that the problem can be solved only if a significant amount of additional a priory information is introduced by the researcher.
To simplify structure elucidation of this complex molecule we utilized the fact that spectra of
shermilamines D and E are similar to those of cycloshermilamine D. Therefore we introduced a user defined fragment 2 existing in known structures of the given series.
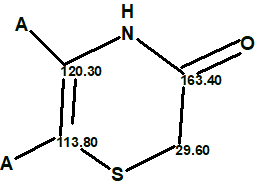
2
This fragment was manually drawn in the MCD (Figure 2).
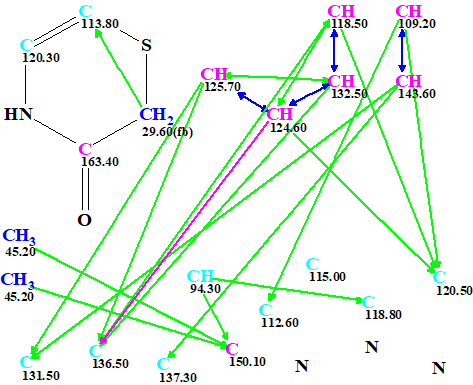
Figure 2. Edited MCD.
Structure generation was initiated from the edited MCD with the constraint that only 5- and 6-membered rings were allowed. Results: k = 217040 → 564 → 263, tg = 3 m 50 s.
According to the general CASE strategy, prediction of 13C chemical shifts was performed by three empirical methods implemented into ACD/Structure Elucidator (HOSE code based, incremental and neural networks) and the output structural file was ranked in increasing order of average deviations dA(13C) calculated using HOSE codes. The four top ranked structures are presented in Figure 3.
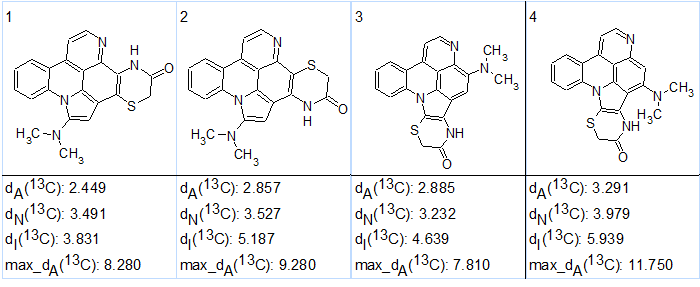
Figure 3. The four top ranked structures of the output file.
Figure 3 shows that the best structure #1 coincides with the structure 1 suggested in the article [1], while a very similar structure was placed in the second position. We see that the next two isomers, 2 and 3, have very similar sets of carbon chemical shift deviations, and thus should be considered as plausible candidates. Therefore the ranking which was carried out on the basis of empirical methods of NMR chemical shift prediction has been checked [2] using the quantum-chemical approach methodology suggested in [3].
DFT calculations of carbon chemical shifts for the four CASE suggested structures of cycloshermilamine D were done at the mPW1PW91/6-311+G(2d,p) level of theory with inclusion of the polarizable continuum model for chloroform (scrf = (solvent =chloroform, smd)). Carbon chemical shift values were then derived from calculated isotropic shielding constants by applying the following scaling coefficients: 186.5242 (intercept) and −1.0533 (slope) [4]. Prior to DFT chemical shift calculations the molecular geometries were optimized at the B3LYP/6-31+G(d,p) level of DFT. Unless otherwise stated, all DFT calculations were carried out by the Gaussian09 software package [5]. The experimental and DFT calculated carbon chemical shifts for the four suggested isomers of cycloshermilamine D by CASE analysis are shown in Table 2.
Table 2. Results of DFT calculations.
| Carbons | Exp. | #1 | #2 | #3 | #4 |
| 1 | 143.6 | 145.22 | 146.06 | 143.49 | 146.91 |
| 2 | 109.2 | 108.05 | 106.54 | 107.87 | 107.32 |
| 3 | 131.5 | 131.83 | 131.35 | 131.91 | 130.10 |
| 4 | 120.5 | 120.46 | 121.04 | 120.32 | 120.78 |
| 5 | 125.7 | 125.13 | 124.75 | 125.03 | 125.48 |
| 6 | 124.6 | 122.65 | 122.70 | 122.14 | 122.48 |
| 7 | 132.5 | 130.09 | 129.56 | 129.46 | 129.30 |
| 8 | 118.5 | 117.40 | 117.16 | 115.19 | 115.21 |
| 9 | 136.5 | 135.63 | 135.32 | 134.32 | 134.69 |
| 10 | 150.1 | 147.83 | 147.73 | 143.53 | 149.28 |
| 11 | 94.3 | 94.54 | 91.24 | 103.59 | 107.86 |
| 12 | 115.0 | 112.82 | 106.74 | 106.69 | 104.07 |
| 13 | 113.8 | 114.19 | 109.86 | 109.45 | 109.56 |
| 14 | 29.6 | 33.82 | 33.96 | 36.54 | 35.94 |
| 15 | 163.4 | 161.06 | 162.73 | 160.78 | 159.81 |
| 16 | 120.3 | 124.48 | 129.33 | 120.88 | 121.66 |
| 17 | 137.3 | 132.54 | 141.53 | 139.81 | 146.43 |
| 18 | 112.6 | 109.57 | 108.68 | 111.88 | 108.54 |
| 19 | 118.8 | 117.43 | 121.42 | 119.50 | 126.16 |
| NMe-1 | 45.2 | 43.29 | 43.32 | 40.42 | 45.07 |
| NMe-2 | 45.2 | 41.20 | 41.14 | 41.04 | 39.75 |
| RMSD, ppm | – | 2.39 | 3.71 | 4.10 | 5.37 |
| max_d, ppm | – | 4.76 | 9.03 | 9.29 | 13.56 |
The DFT analysis of chemical shifts clearly shows that the first structure has the lowest RMSD and max_d, consistent with the earlier proposed assignment. The effect of the sulfur heavy atom on adjacent carbon (C13) is minimal (113.8 ppm experimental vs 114.2 ppm calculated).
Structure 1 together with the 13C chemical shift assignments determined by the program is shown below.
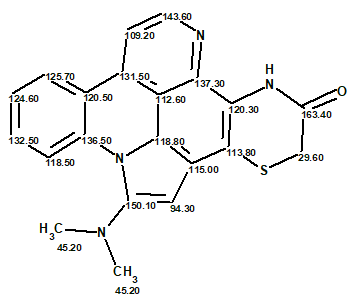
This is another case where the combination of CASE and DFT chemical shift analysis credibly supported the published structure of cycloshermilamine D without any additional experimental data.
References
- G. Koren-Goldshlager, M. Aknin, Y. Kashman. (2000). Cycloshermilamine D, a New Pyridoacridine from the Marine Tunicate Cystodytes violatinctus. J. Nat. Prod., 63: 830–831.
- A.V. Buevich, M.E. Elyashberg. (2018). Towards unbiased and more versatile NMR-based structure elucidation: A powerful combination of CASE algorithms and DFT calculations. Magn. Reson. Chem., 56, 493–504.
- A.V. Buevich, M.E. Elyashberg. (2016). Synergistic combination of CASE algorithms and DFT chemical shift predictions: a powerful approach for structure elucidation, verification and revision. J. Nat. Prod., 79(12): pp 3105–3116.
- Chemical Shift Repository for computed NMR scaling factors, with Coupling Constants Added Too. Retrieved June 27, 2017 from http://cheshirenmr.info.
- M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, et al. (2009). Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013.
About the Author
