April 1, 2021
by Mikhail Elyashberg, Leading Researcher, ACD/Labs
Damirine B
The indolocarbazole family of bisindole alkaloids is a well-known class of natural products. It’s best-known member is staurosporine, a protein kinase C inhibitor that belongs to the indolo[2,3‐a] carbazole structural class. Several other indolo[2,3‐a] carbazoles have so far been isolated and identified. However, only a limited number of other isomeric forms of indolocarbazole has been reported. Gustafson et al [1] investigated an extract of the marine sponge Damiria sp., which belongs to an understudied genus. Two novel alkaloids, named damirines A and B, were isolated as part of this. Their structures were elucidated using comprehensive NMR spectroscopic studies which, for the case of damirine B (structure 1), included LR‐HSQMBC spectra. This is a long-range heteronuclear correlation experiment that is optimized to work for very low coupling constants, thus being very useful for proton deficient structures.
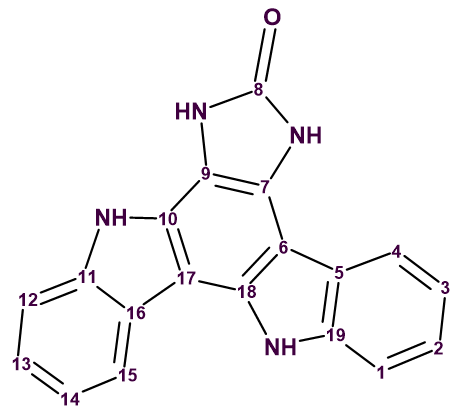
1
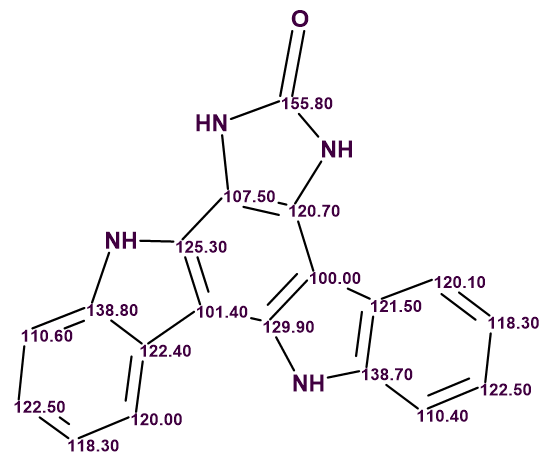
Damirine B (2) was isolated as a brown powder with a molecular formula found by (+)‐HR-ESI-MS measurements to be C19H12N4O. The ratio of skeletal atoms to hydrogens is 2, indicating a highly hydrogen-deficient molecule. For elucidating the structure of it, together with 1H, 13C, 1H-1H COSY, 1H-13C HSQC, 1H-13C HMBC spectra, 1H-15N HSQC, ROESY and LR-HSQMBC spectra were recorded.
The key correlations observed in 2D NMR spectra by the authors [1] are shown in Figure 1.
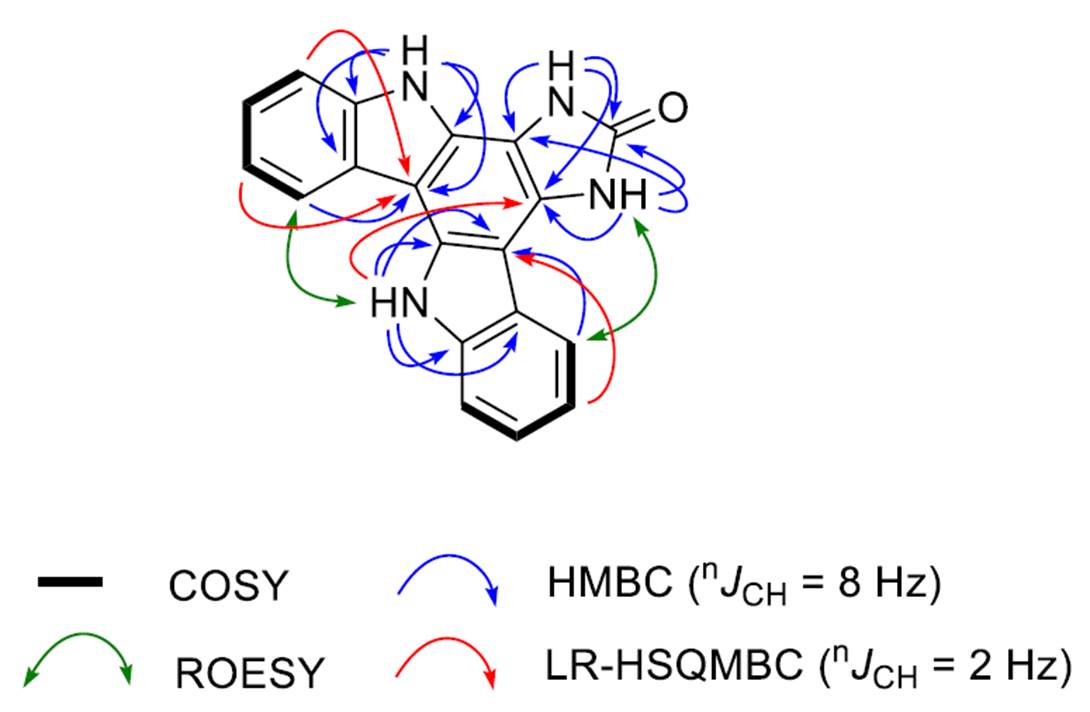
Figure 1. Key correlations observed in 2D NMR spectra of damirine B.
It was interesting to find out if such a challenging structure could be elucidated using ACD/Structure Elucidator without using LR-HSQMBC. Therefore, we entered the spectroscopic data presented in Table 1 to ACD/SE.
Table 1. Spectroscopic data entered to ACD/SE.
| C/X Label | δC | δC calc (HOSE) | XHn | δH | COSY | H to C HMBC |
| C 1 | 110.400 | 110.920 | CH | 7.610 | 7.30 | C 3, C 5 |
| C 2 | 122.500 | 125.630 | CH | 7.300 | 7.16, 7.61 | C 1, C 4, C 19 |
| C 3 | 118.300 | 119.270 | CH | 7.160 | 7.30, 8.48 | C 1, C 5 |
| C 4 | 120.100 | 118.170 | CH | 8.480 | 7.16 | C 6, C 2, C 19 |
| C 5 | 121.500 | 124.090 | C | |||
| C 6 | 100.000 | 105.230 | C | |||
| C 7 | 120.700 | 115.910 | C | |||
| C 8 | 155.800 | 155.680 | C | |||
| C 9 | 107.500 | 111.340 | C | |||
| C 10 | 125.300 | 130.860 | C | |||
| C 11 | 138.800 | 139.210 | C | |||
| C 12 | 110.600 | 110.920 | CH | 7.550 | 7.32 | C 14, C 16 |
| C 13 | 122.500 | 124.600 | CH | 7.320 | 7.23, 7.55 | C 12, C 15, C 11 |
| C 14 | 118.300 | 117.980 | CH | 7.230 | 7.32, 8.56 | C 12, C 16 |
| C 15 | 120.000 | 119.380 | CH | 8.560 | 7.23 | C 17, C 13, C 11 |
| C 16 | 122.400 | 120.270 | C | |||
| C 17 | 101.400 | 105.610 | C | |||
| C 18 | 129.900 | 131.610 | C | |||
| C 19 | 138.700 | 139.130 | C | |||
| N 1 | NH | 11.320 | C 9, C 7, C 8 | |||
| N 2 | NH | 11.820 | C 9, C 7, C 8 | |||
| N 3 | NH | 12.730 | C 17, C 16, C 10, C 11 | |||
| N 4 | NH | 11.560 | C 6, C 5, C 18, C 19 |
The Molecular Connectivity Diagram (MCD) created automatically by the program is shown in Figure 2.
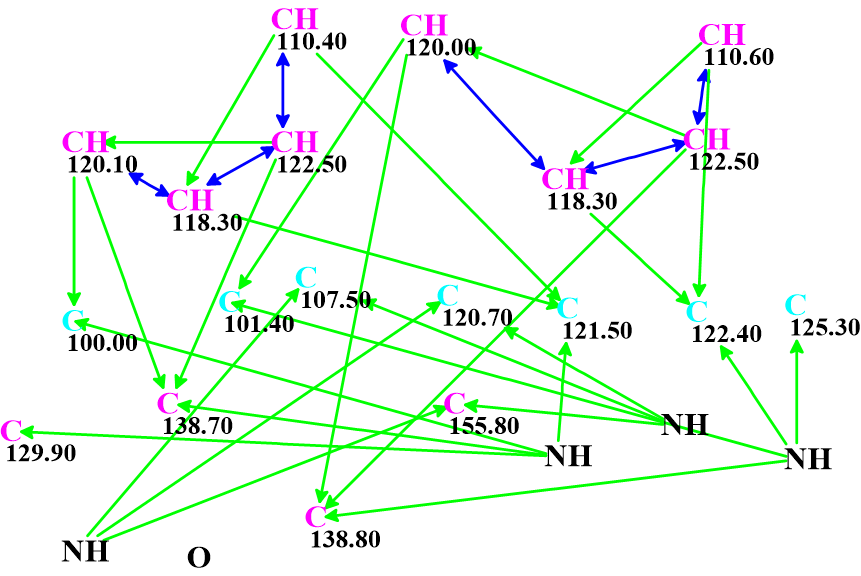
Figure 2. Molecular Connectivity Diagram.
As is the case, green and blue arrows indicate HMBC and COSY correlations respectively (2-3 bonds and 1-bond away). The carbon atoms for which sp2 hybridization was automatically assigned by the program are shown in violet, while atoms assigned to be not sp (sp3 or sp2) are light blue.
No manual edits to the MCD were made. Checking the MCD for consistency showed that no contradictions were detected, and strict structure generation followed by 13C chemical shift prediction and spectral filtering was initiated. Result: k = 133,526 → (filtering) → 28 → (duplicate removal) → 6. The time required was tg = 1 min 10 sec. Adding the LR-HSQMBC correlations gave exactly the same final results in terms of number of structures and computation time, indicating that the problem was already sufficiently constrained with the existing data.
The standard procedure for ACD/SE 13C chemical shift prediction was followed, using three algorithms (Incremental, HOSE codes and Neural Network). The structures were ranked in increasing order of average deviations. The three top ranked structures are shown in Figure 3.
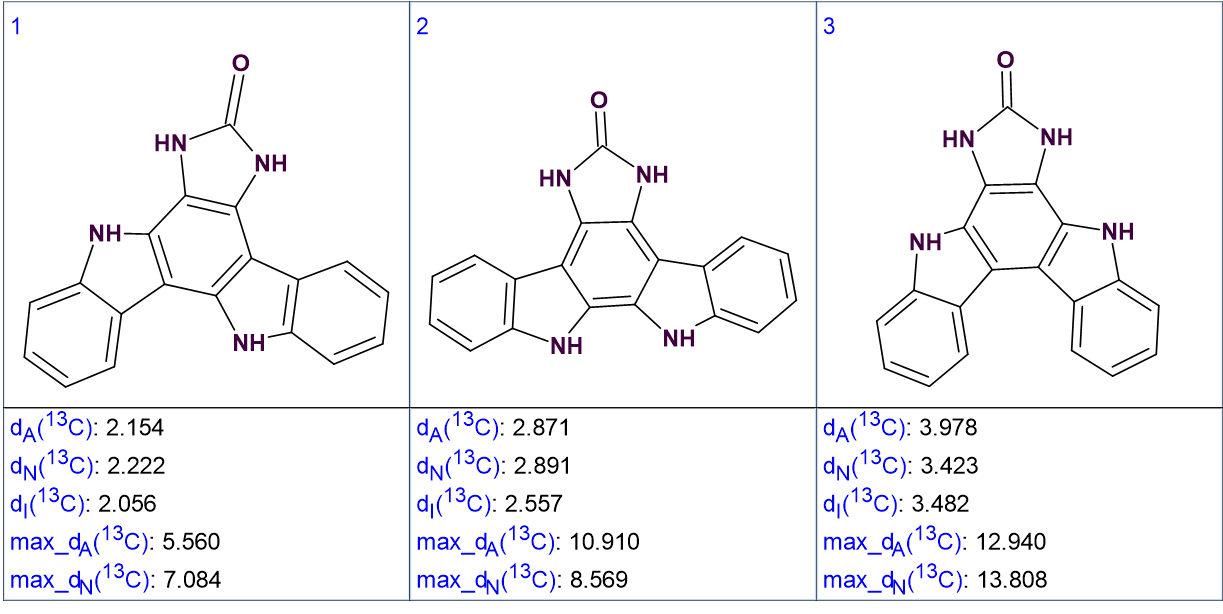
Figure 3. The three top ranked structures of the output file.
We see that the best structure coincides with the structure of damirine B determined by the authors of [1]. As the average deviations calculated for structures #1 and #2 do not differ significantly it is desirable to confirm the validity of the first structure by some other means. For this reason, the correlations obtained by ROESY can be used (see Figure 4).
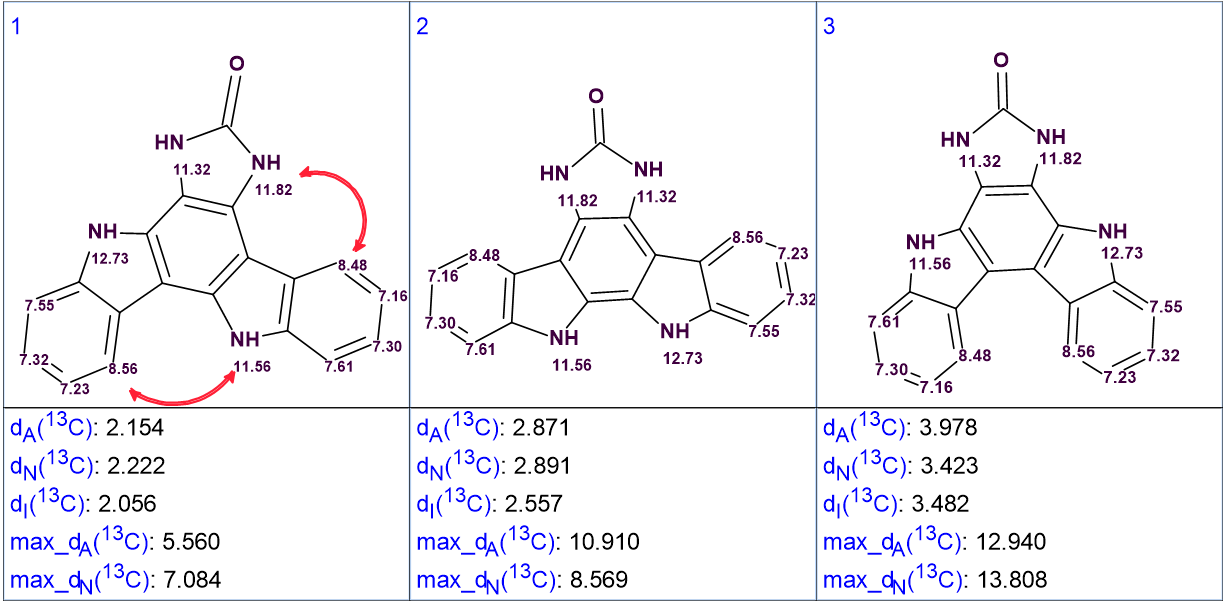
Figure 4. The top ranked structures with assigned 1H chemical shifts. Red arrows show the ROESY correlations observed by the authors [1].
It is evident that the ROESY correlation between the protons with signals at 8.56 ppm and 11.56 ppm could not be possible in structure #2, which further supports the validity of the solution. Structure #1 with automatically assigned chemical shifts is shown below:
In conclusion, we showed that the correct structure of a hydrogen deficient molecule was elucidated fully automatically in a minute without using LR-HSQMBC data. Nevertheless, the LR-HSQMBC experiments has been shown to be very helpful in solving other structures, so its value should not be questioned.
References:
- T. D. Tran, L. K. Cartner, H. R. Bokesch, C. J. Henrich, X.W. Wang, C. Mahidol, S. Ruchirawat, P. Kittakoop, B.R. O’Keefe, K. R. Gustafson. (2021). NMR characterization of rearranged staurosporine aglycone analogues from the marine sponge Damiria sp. Magn. Reson. Chem., 59: 534-539.
About the Author
