May 1, 2012
by Mikhail Elyashberg, Leading Researcher, ACD/Labs
Lycojaponicumin
Recently Wang et al1 isolated a novel alkaloid lycojaponicumin (C16H21NO4, structure 1) from the alkaloidal extract in a trace amount, with a unique 5/5/5/5/6 pentacyclic ring system including two fused tetrahydroisoxazole rings. The structure was remarkable for its unprecedented skeleton formed by new C-C bond linkage, which had never existed in Lycopodium alkaloids. Furthermore, the nitrogen atom in structure 1 was attached to C(88.00) through an oxygen atom to form a 1-aza-7-oxabicyclo[2,2,1]heptane moiety, which was first reported in natural products. The structure was elucidated by spectroscopic methods and X-ray diffraction analysis.
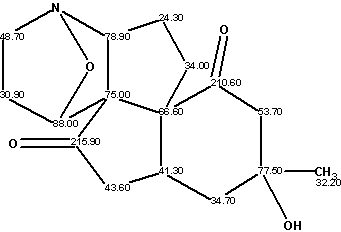
Structure 1
From a spectroscopic point of view, the structure of this unusual alkaloid contains two carbon atoms—C(75.00) and C(66.60)—whose chemical shifts are characteristic for carbons having a neighbor heteroatom (most likely oxygen) in the first sphere environment.2 These chemical shifts are rarely observed for atoms with a carbon environment. In addition, the chemical shift at 78.9 ppm is typical for a carbon atom containing an oxygen in the first sphere (not a nitrogen atom). All possible environments must be tried for the mentioned carbon atoms during the structure elucidation. A comprehensive search of all allowed atomic combinations would obviously take a lot of human effort if the structure elucidation was performed manually.
In the original publication,1 spectroscopic data were presented by tables of 1D NMR 13C and 1H chemical shifts, while COSY and HMBC correlations were graphically depicted on structure 1 below:
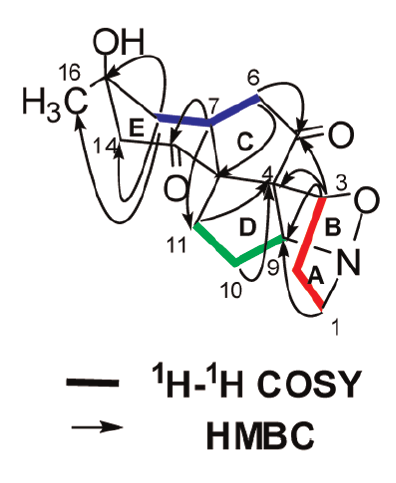
COSY and HMBC correlations observed for structure 1.
1D and 2D NMR data were input in Structure Elucidator and a MCD (Molecular Connectivity Diagram) was automatically created by the program. An option “Allow sp carbons” was switched off, as no features of triple bonds were observed in the IR spectrum.
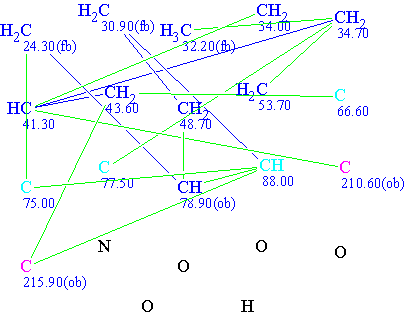
Molecular Connectivity Diagram (MCD) for lycojaponicumin based on the published NMR data. “ob” indicates that the neighboring heteroatom is set by the program as obligatory; “fb” indicates forbidden.
Carbon atoms with chemical shifts 66.6, 75.5, 77.5, and 88.0 are colored in light blue in the MCD, which means that their hybridization states can be either sp2 or sp3. No suggestions on the possibility of having neighboring heteroatoms are shown—the possibility is determined by the program during the structure generation.
First run
For the first run, bonds between heteroatoms were forbidden because such bonds are relatively rare in molecules of natural products. Experience shows that when chemical bonds between heteroatoms are allowed, the number of generated structures frequently increases dramatically. Therefore the first attempt to solve a problem is usually made under the assumption that bonds between heteroatoms are absent in the unknown molecule (“try your luck” mode). The following results were obtained: k=7144→3307→2889, tg=8.3s.
13C chemical shift calculation for the output file and structure ranking revealed that the “best” structure was characterized by deviation values which were too large (5–6 ppm) which allows one to conclude that the solution is most likely incorrect.
The following reasons for an incorrect solution can be mentioned: a) presence of nonstandard correlations (nJHH,CH>3) in 2D NMR data, b) wrong suggestions (“axioms”) on atom properties were used, c) the correct structure did not pass spectral or structural filtering, d) suggestions on the absence of chemical bonds between heteroatoms is wrong. In our case, checking the MCD for the presence of nonstandard connectivities in the 2D NMR data showed that all of them were of standard length. No user corrections of MCD and atom properties were made, so incorrect user suggestions on atom properties is not at fault. Rejection of the correct structure by the filtering procedure happens very rarely and thus is also likely not the culprit.
Second run
Therefore the second attempt of structure generation can be made using options where chemical bonds between heteroatoms are allowed. Structure generation was accompanied with 13C chemical shift prediction for the generated structure and structures for which d>4 ppm were rejected. Results: k=16246→1, tg=52s. Only a single structure was obtained, structure 1, with a deviation value dI=3.8 ppm.
Taking into account that the average deviation characteristic for empirical methods of 13C chemical shift prediction is ca. 1.6–1.8 ppm, the deviation calculated for structure 1 is large enough, which is explained by a very unusual structure of the new natural product. Repeated structure generation with a switched off filter confirmed that structure 1 has smallest deviations and is indeed the best one.
Third run
To be fully sure that the proposed structure is the right one, structure generation was repeated with the Atomic Properties Correlation Table (APCT) switched off. This table is used not only for automatic setting of atom properties during MCD creation, but for preventing the generation of incorrect structures (those that contain carbons with improbable environments) in the process of structure generation. The following result was obtained: k=2,011,349→1,941,788→1, tg=1 h 08 m 37 s, and the single output structure coincided with structure 1.
This example clearly shows the high efficiency of utilizing APCT during the structure generation—the time to perform structure generation was ca. 4000 times longer when APCT was switched off.
It was interesting to see what would happen if we suggested that carbons C(75.00) and C(66.60) have a heteroatom in the first sphere of the environment (this suggestion is the most probable one according to known characteristic spectral features2). When a property sp3/ob was set for the atom C(75.00), the structure generation accompanied with 13C chemical shift prediction gave the following result: k=6399→0, tg=23s. A similar result was obtained for C(66.6): k=9798→0, tg=33s. Thus, both wrong hypotheses (“axioms”) were immediately rejected by the program. Note that it would take a significant amount of time for a chemist to check these hypotheses if a manual approach for structure elucidation were used.
In conclusion, an unprecedented structure of a novel alkaloid was elucidated by the system almost automatically. No user suggestions were used. A hint for setting more free options of structure generation, when the existence of chemical bonds between heteroatoms is allowed, was given by the system itself.
References
- Lycojaponicumins A – C, Three Alkaloids with an Unprecedented Skeleton from Lycopodium japonicum. X.-J.Wang, G.-J. Zhang, P.-Y. Zhuang, Y. Zhang, S.-S. Yu, X.-Q. Bao, D. Zhang, Y.-H. Yuan, N.-H. Chen, S.-G. Ma, J. Qu, Y. Li. Org. Lett. ASAP, Received April 12, 2012
- Pretsch, E.; Clerc, T.; Seibl, J.; Simon, W., Tables of Spectral Data for Structure Determination of Organic Compounds. Springer-Verlag: Berlin, 1989.
About the Author
