October 1, 2020
by Mikhail Elyashberg, Leading Researcher, ACD/Labs
Melohemsine J
The plants of the genus Melodinus (Apocynaceae) include 53 species and several of them have been used in traditional Chinese medicine. They can be found in regions of southern China and, together with the Loganiaceae and Rubiaceae families they are known to contain many monoterpenoid indole alkaloids. For these reasons they have been studied a lot by chemists and pharmacologists. Zhang et al [1], while studying species of the Melodimus genera isolated and elucidated a new alkaloid, melohemsine J (1).
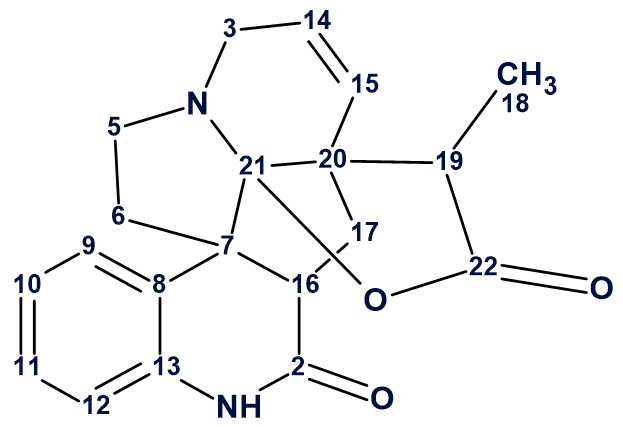
1
Melohemsine J (1) is the first melodinus-type alkaloid featuring a 6/6/5/5/ 6/5 ring skeleton and a tetrahydrofuro[2,3-b]pyridine-2(3H)-one unit. The new compound and its absolute configuration were characterized by spectroscopic methods, X-ray diffraction data, and computational methods.
Melohemsine J (1) was obtained as colorless crystals from MeOH. Its molecular formula, C20H20N2O3, was determined from the 13C NMR data and protonated ion [M + H]+ at m/z 337.1543 (calcd for C20H21N2O3: 337.1547) in its HRESIMS spectrum. Its IR spectrum (KBr disk, Figure 1) displayed the presence of amino (weak band at 3206 cm−1) and supposedly two carbonyl (1770, 1673 cm−1) groups. The first band, 1770 cm−1, is characteristic for a 5-membered lactone, while the second, 1673 cm−1, most probably for an amide group.
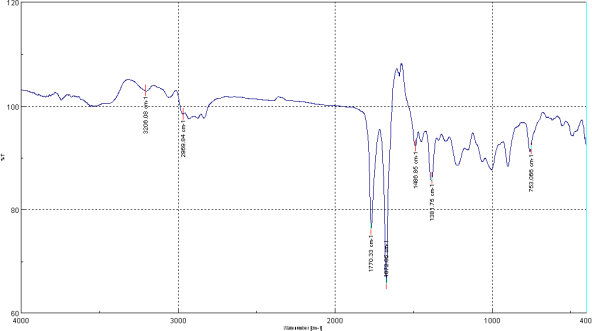
Figure 1. IR spectrum of Melohemsine J (KBr disk).
To determine the structure of the new compound, authors [1] utilized 1D and standard set of 2D NMR spectra – HSQC, HMBC and COSY. It should be noted that only key HMBC correlations wee presented in the article [1]. These spectroscopic data (Table 1) were entered into ACD/Structure Elucidator software and used for challenging the program.
Table 1. NMR spectroscopic data.
| Label | δC | СHn | H | M | COSY | H to C HMBC |
| C 2 | 173.000 | C | ||||
| C 3 | 43.400 | CH2 | 3.430 | u | ||
| C 3 | 43.400 | CH2 | 3.340 | u | 6.04 | C 5, C 21 |
| C 5 | 50.700 | CH2 | 3.310 | u | 2.15 | C 7, C 21 |
| C 5 | 50.700 | CH2 | 3.430 | u | ||
| C 6 | 38.800 | CH2 | 2.310 | u | ||
| C 6 | 38.800 | CH2 | 2.150 | u | 3.31 | C 7, C 21 |
| C 7 | 59.200 | C | ||||
| C 8 | 124.300 | C | ||||
| C 9 | 130.200 | CH | 7.620 | d | 7.06 | C 7 |
| C 10 | 124.900 | CH | 7.060 | t | 7.24, 7.62 | |
| C 11 | 129.700 | CH | 7.240 | t | 6.91, 7.06 | C 13 |
| C 12 | 117.100 | CH | 6.910 | d | 7.24 | |
| C 13 | 137.700 | C | ||||
| C 14 | 126.800 | CH | 6.040 | u | 3.34, 5.76 | |
| C 15 | 126.100 | CH | 5.760 | d | 6.04 | C 21 |
| C 16 | 50.800 | CH | 3.100 | t | 2.23 | |
| C 17 | 43.100 | CH2 | 2.330 | u | ||
| C 17 | 43.100 | CH2 | 2.230 | u | 3.10 | C 7, C 21, C 2 |
| C 18 | 10.000 | CH3 | 1.010 | d | 2.14 | C 20, C 22 |
| C 19 | 38.700 | CH | 2.140 | u | 1.01 | C 17 |
| C 20 | 49.600 | C | ||||
| C 21 | 113.400 | C | ||||
| C 22 | 179.200 | C |
The Molecular Connectivity Diagram (MCD) automatically produced by the program from the data collected in Table 1 is shown in Figure 2.
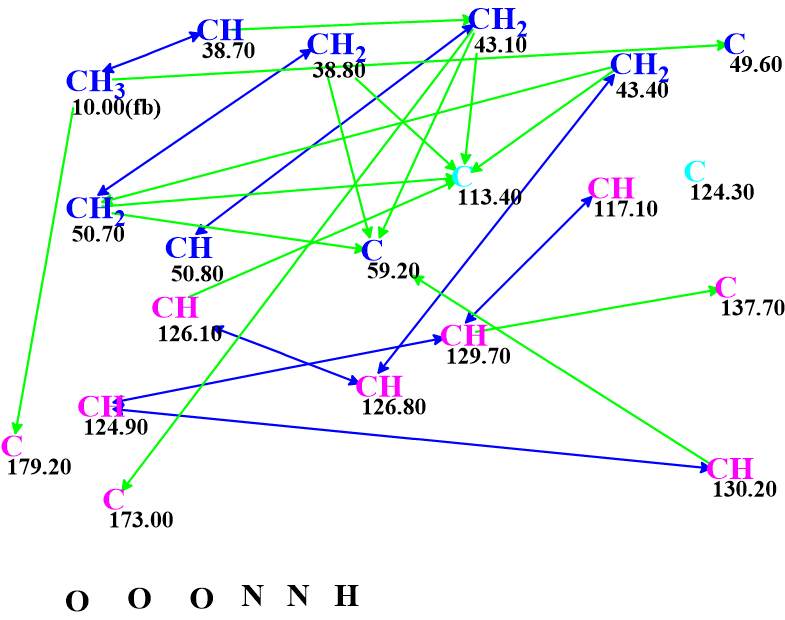
Figure 2. Molecular Connectivity Diagram.
No manual edits were done to the MCD, and structure generation accompanied with fast 13C chemical shift prediction by an incremental method and spectral filtering were initiated from the constraints summarized. The generation gave the following results: k = 135,893 → (spectral filtering) → 35 → (removing duplicates) → 35, tg = 3 m 12 s.
Then, 13C chemical shift prediction by all three methods common for ACD/Structure Elucidator (beside of the mentioned incremental method, HOSE code based and neural network approaches) was carried out. The next step was structural file ranking in descending order of dA (13C) value (an average deviation of HOSE calculated 13C chemical shifts from experimental ones) were performed. 1H chemical shifts were also calculated. The five top ranked structures are presented in Figure 3.
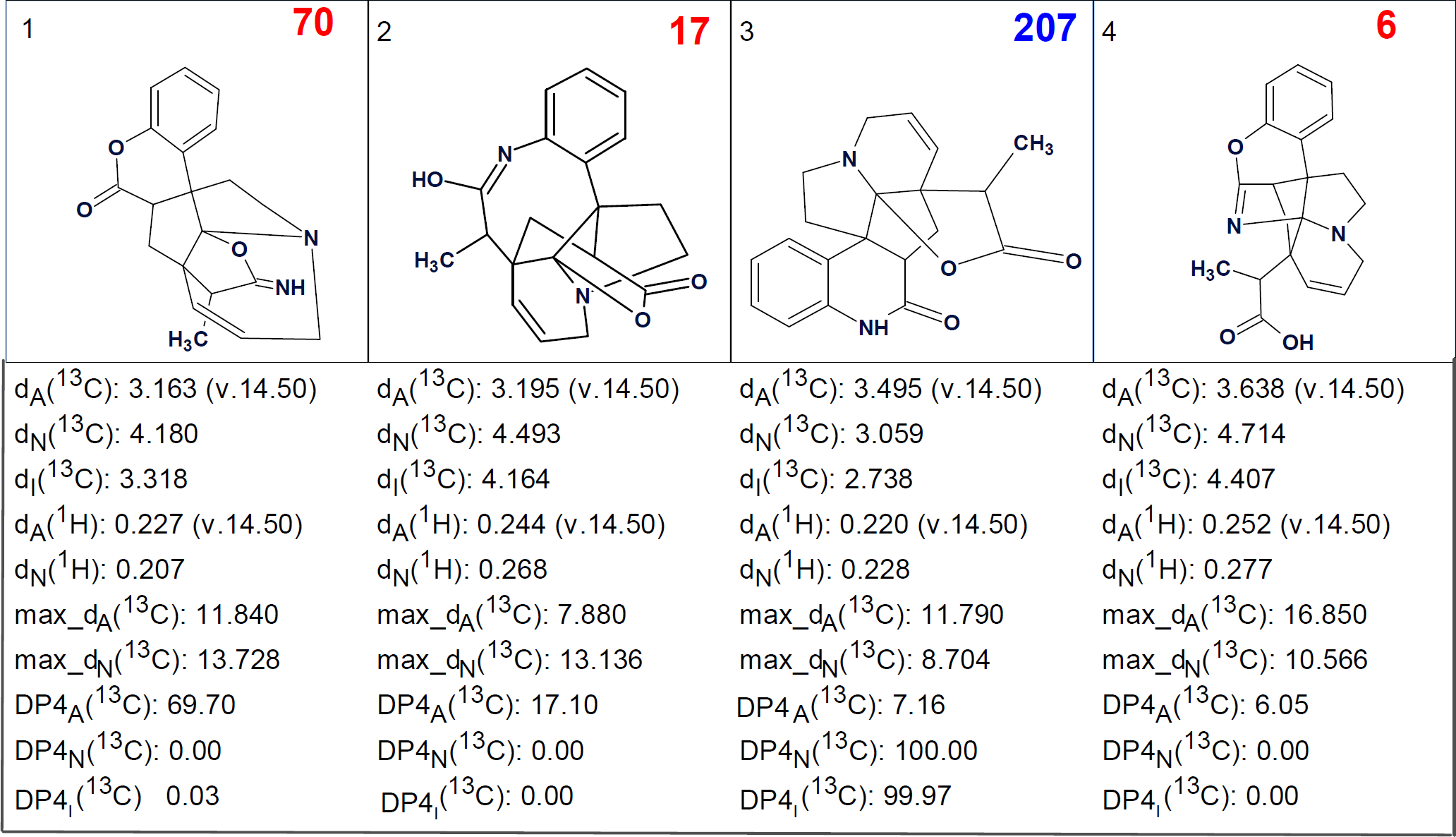
Figure 3. Five top ranked structures of the output file.
We see that structure of melohemsine J determined by authors [1] was generated and placed by the ranking procedure at the third position. This can be explained by the unusual polycyclic structure of the molecule under investigation, which led to relatively high values of deviations calculated by all methods available. In addition, the values of dA(13C) deviations are very close. Though the HOSE code-based method of NMR chemical shift prediction usually ranks structures more accurately than the other two, in this case minimum deviations were delivered by calculations performed using increments and neural networks.
In v2020.1 of ACD/Structure Elucidator a new method of structure probability calculation similar to the well-known DP4 approach [2], has been included. DP4 was developed for the analogous case when the DFT chemical shift predictions for a given set of structure are very close. By using a multiplicative probability together with the relative errors in each prediction DP4 can give a more definite result in many cases. Since we are testing this approach with many of the problems we have already solved, we decided to apply it to the solution presented in Figure 3. The values of DP4A (13C), DP4N (13C), DP4I (13C) are shown for all structures.
The probabilities DP4A (13C), DP4N (13C), DP4I (13C) are calculated based on different algorithms for 13C chemical shifts prediction, while the error distributions for each method are different. Therefore, it can be assumed that the values DP4A (13C), DP4N (13C), DP4I (13C) are independent and they can be considered as the number of votes or the number of points awarded to a given structure by a given method. Consequently, we can use a value equal to the sum of the corresponding probabilities as a score characterizing general assessment of the reliability of a given structure. These values are shown along with structures in corresponding cases (Figure 3). We see that the correct structure #3 received the highest score 207, which allows us to declare it the most probable. It is obvious that structures #2 and #4, having score factors 17 and 6 correspondingly, can be rejected because they contain OH group whose presence in the molecule would have been evident from the IR spectrum (a strong absorption band at 3000-3200 cm-1 is absent from IR spectrum, Figure 1). However, it is probably possible to verify the validity of structure #3 if one has the full set of observed HMBC correlations and not just the key ones, as the authors have published. It is quite common that once all the correlations are utilized then some of the structures are not generated at all. Alternatively, DFT based 13C chemical shift calculations for structures #1 and #3 (not for all 4 structures) could also provide an answer.
Structure #3 with assigned 13C chemical shifts is shown below.
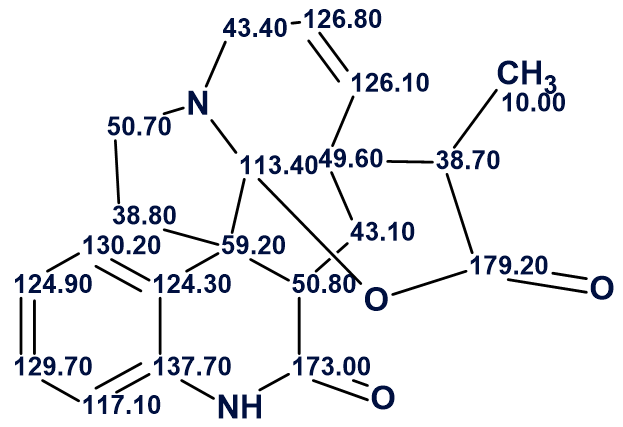
Thus, the structure of a new alkaloid having unusual skeleton was automatically elucidated with the use of ACD/Structure Elucidator.
References
- J. Zhang, Z.-W. Liu, Y. Li, C.-J. Wei, J. Xie, M.-F. Yuan, D.-M. Zhang, W.-C. Ye, X.-Q. Zhang. (2020). Structurally Diverse Indole Alkaloids with Vasorelaxant Activity from Melodinus hemsleyanus. Nat. Prod., 83: 2313−2319.
- S.G. Smith, J.M. Goodman. (2010). Assigning Stereochemistry to Single Diastereoisomers by GIAO NMR Calculation: The DP4 Probability. J. Am. Chem. Soc., 132: 12946-12959.
- A. V. Buevich, M. E. Elyashberg. (2016). Synergistic combination of CASE algorithms and DFT chemical shift predictions: a powerful approach for structure elucidation, verification and revision. J. Nat. Prod., 79(12): 3105–3116.
- A.V. Buevich, M. E. Elyashberg. (2018). Towards unbiased and more versatile NMR-based structure elucidation: A powerful combination of CASE algorithms and DFT calculations. Magn. Reson. Chem., 56: 493–504. DOI: 10.1002/mrc.4645
- A.V. Buevich, M.E. Elyashberg. (2020). Enhancing Computer Assisted Structure Elucidation with DFT analysis of J-couplings. Magn. Reson. Chem., 58(6): 594-606. DOI: 10.1002/mrc.4996
About the Author
