August 1, 2021
by Mikhail Elyashberg, Leading Researcher, ACD/Labs
Trichilianone D
Trichilia species, a genus of plants from the Meliaceae family, are found in tropical forests and are used in traditional medicine. This species is poorly investigated and so a phytochemical study was published just recently [1]. This included the isolation of metabolites and the testing for antileishmanial activity.
The study was focused on the characterization of metabolites present in an extract of the bark of Trichilia adolfi. In particular, the structure of the isolated metabolite Trichilianone D (1) was elucidated after an extensive spectroscopic analysis based essentially on the elemental composition determined by high-resolution mass spectrometry and 1D NMR as well as 2D NMR experiments. Trichilianone D is, through HRMS experiments, suggested to have the elemental composition C29H34O11 (m/z: 559.2171 [M + H]+, calculated for C29H35O11 559.2179).
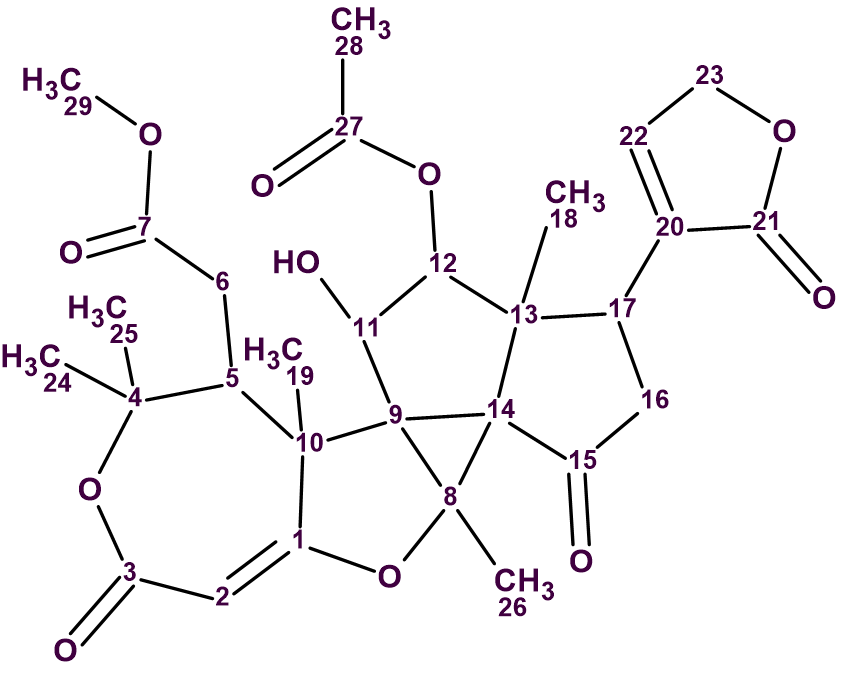
1
The 1D and 2D NMR spectroscopic data which were used in [1] for trichilianone D structure elucidation are presented in Table 1.
Table 1. 1D and 2D NMR spectroscopic data.
| Label | δC | δC calc (HOSE) | XHn | δH | M(J) | COSY | H to C HMBC |
| C 1 | 180.400 | 177.330 | C | ||||
| C 2 | 94.400 | 92.700 | CH | 5.500 | u | C 10, C 3, C 1 |
|
| C 3 | 168.600 | 167.100 | C | ||||
| C 4 | 83.100 | 83.010 | C | ||||
| C 5 | 49.800 | 40.910 | CH | 2.600 | u | 3.82 | |
| C 6 | 35.200 | 34.540 | CH2 | 3.820 | u | 2.60 | C 5, C 10, C 4, C 7 |
| C 6 | 35.200 | 34.540 | CH2 | 2.780 | u | ||
| C 7 | 174.700 | 173.150 | C | ||||
| C 8 | 77.600 | 76.110 | C | ||||
| C 9 | 58.000 | 55.990 | C | ||||
| C 10 | 56.200 | 51.720 | C | ||||
| C 11 | 82.600 | 79.510 | CH | 4.340 | u | 3.76, 5.62 |
C 9, C 8 |
| C 12 | 91.600 | 87.870 | CH | 5.620 | u | 4.34 | C 9, C 27 |
| C 13 | 53.700 | 44.470 | C | ||||
| C 14 | 58.900 | 56.680 | C | ||||
| C 15 | 208.500 | 209.150 | C | ||||
| C 16 | 43.300 | 43.230 | CH2 | 2.760 | u | ||
| C 16 | 43.300 | 43.230 | CH2 | 3.110 | u | 3.36 | C 17, C 14, C 20, C 15 |
| C 17 | 42.300 | 44.220 | CH | 3.360 | u | 3.11 | C 20 |
| C 18 | 13.400 | 18.360 | CH3 | 0.970 | s | C 17, C 13, C 14, C 12 |
|
| C 19 | 19.300 | 14.750 | CH3 | 1.690 | s | C 5, C 10, C 9, C 1 |
|
| C 20 | 132.000 | 133.560 | C | ||||
| C 21 | 173.300 | 174.060 | C | ||||
| C 22 | 148.700 | 148.690 | CH | 7.320 | u | 4.93 | C 17, C 23, C 20, C 21 |
| C 23 | 70.600 | 70.760 | CH2 | 4.930 | u | 7.32 | C 20, C 22, C 21 |
| C 24 | 31.200 | 29.830 | CH3 | 1.460 | s | C 25, C 5, C 4 |
|
| C 25 | 28.600 | 22.490 | CH3 | 1.400 | s | C 24, C 5, C 4, C 3 |
|
| C 26 | 14.600 | 19.290 | CH3 | 1.780 | s | C 9, C 14, C 8, C 15 |
|
| C 27 | 171.900 | 171.560 | C | ||||
| C 28 | 20.800 | 20.930 | CH3 | 2.140 | s | C 27 | |
| C 29 | 52.000 | 51.650 | CH3 | 3.690 | s | C 7 | |
| O 1 | OH | 3.760 | u | 4.34 | C 9, C 11 |
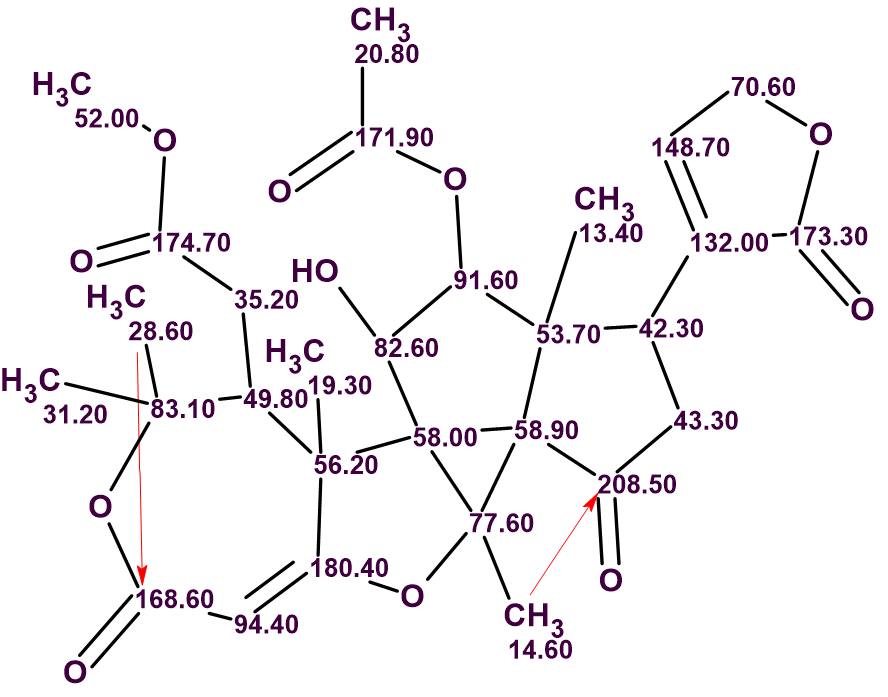
The spectroscopic data were entered into ACD/Structure Elucidator and the program automatically created the Molecular Connectivity Diagram (MCD) shown in Figure 1.
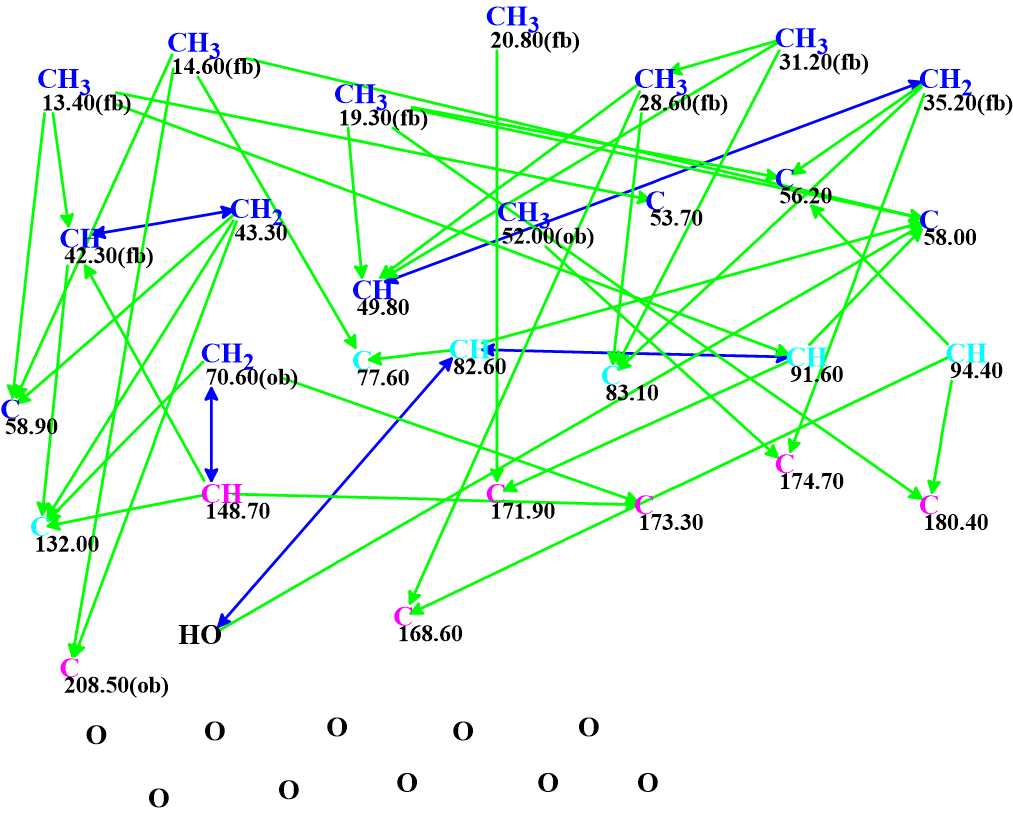
Figure 1. Molecular connectivity diagram. Atom properties were determined by the program automatically.
MCD overview. All carbon atoms colored in blue color were assigned sp3 hybridization. Six light blue carbons C 77.60 – C 132.00 are classified by the program as not sp (sp2 or sp3). Atoms for which neighboring with heteroatoms is forbidden or obligatory are marked by labels “fb” or “ob” correspondingly. HMBC correlations are marked by green arrows, while those of COSY by blue. No manual edits of the MCD were made.
MCD checking showed no contradictions in combined HMBC and COSY data. As the checking procedure is based on heuristic algorithms there is a very small probability that the program has not detected some real contradictions. With this in mind and taking into account the very large number of HMBC and COSY correlations used, Fuzzy Structure Generation accompanied with fast 13C chemical shift prediction was initiated. The generator Options were set automatically. In this mode, the program tries to perform structure generation, first supposing that the number m of non-standard correlations, NSC, is equal to 0. If any structures are generated and written in the output file at m=0, then the generation will stop. Otherwise, the next run of the program will be initiated automatically with m=1. The generation will stop at the m value which provides an output file with structures.
Results: k=168, 905 → (structure filtering) → 48 → (duplicate removing) → 39, tg = 14m 30 s.
2 out of the 45 connectivities have been extended during generation, which means that the output file was produced at m=2.
According to the standard procedure for ACD/Structure Elucidator, 13C chemical shift prediction for all structures was performed using the three methods implemented in the program – HOSE code based approach, Neural Networks and the Incremental method. The chemical shift average deviations dA, dN and dI were calculated, with designations corresponding to the mentioned methods. The structures were ranked in ascending order of average deviations dA.. The six top ranked structures are presented in Figure 2.
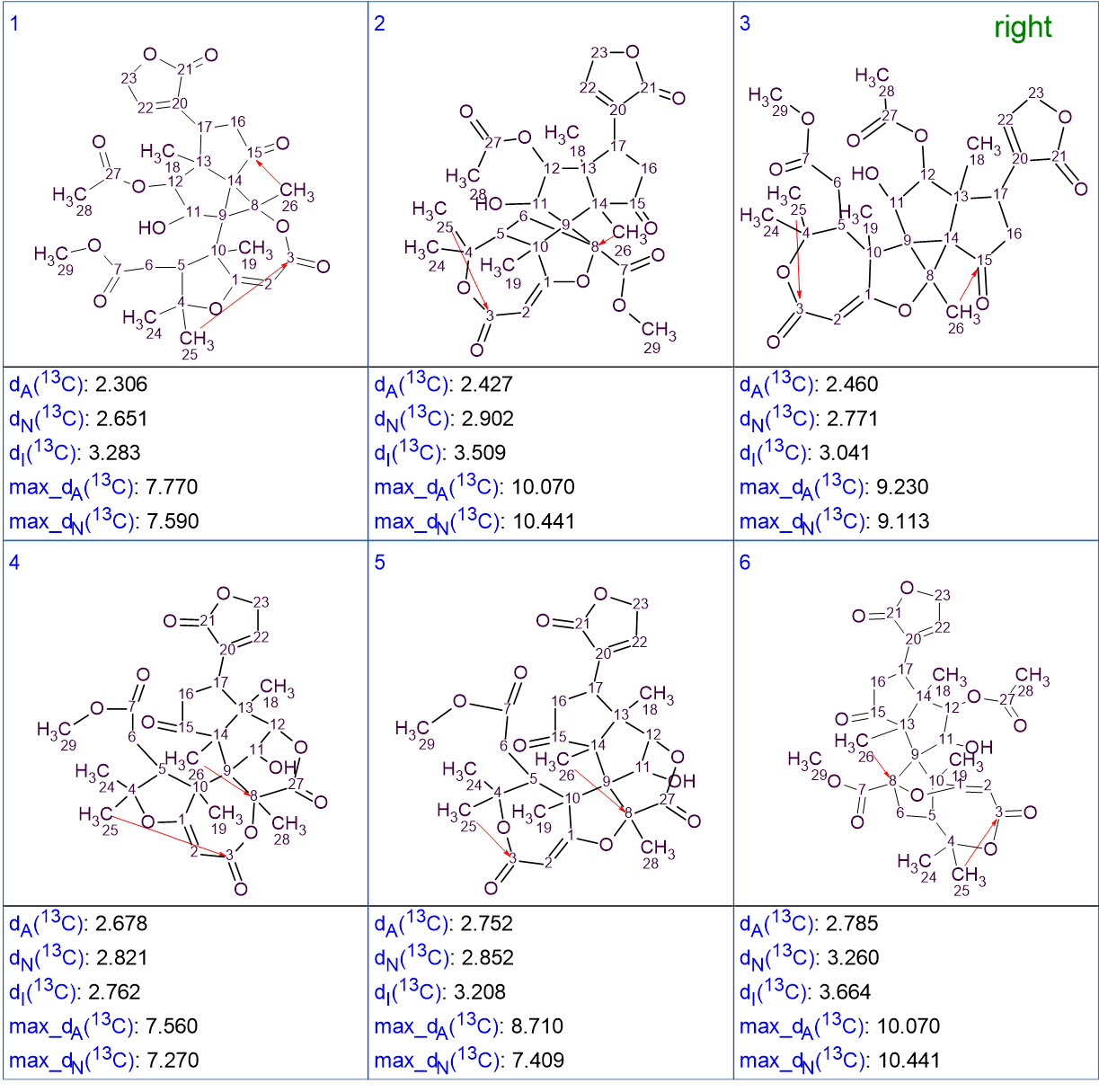
Figure 2. Six top ranked structures. Red arrows show the NSCs detected in HMBC data by the program during Fuzzy Structure Generation.
Comparison of these structures with structure 1 shows that the latter was placed at the third position by the ranking procedure, based on the mean deviations between experimental and predicted chemical shifts. This is not surprising as the values of all deviations are very close. The reason is because of the intrinsic structure similarity amongst the structures. Identifying the correct structure in this case requires a bit more work.
Structures #1 and #4 contain an HMBC correlation that corresponds to a 6JCH. This is highly improbable to be detected with the simple HMBC experiment used by the authors. It could maybe be detected using advanced pulse sequences designed to enhance long range correlations, like the LR-HSQMBC. So, these two structures should be removed from the list.
In order to differentiate between the remaining 4 structures, we can use the DP4 type metrics recently published [2], which use a multiplicative calculation to obtain the probability that a given structure amongst a set is the correct one. Using the DP4 style metrics requires that all the structures be of equal validity, that they are all fully assigned and that we know the chemical shift calculation error for all the shifts. These conditions are all met in this case and ACD/SE supports the calculation of DP4 style metrics. The new results are shown in Figure 3:
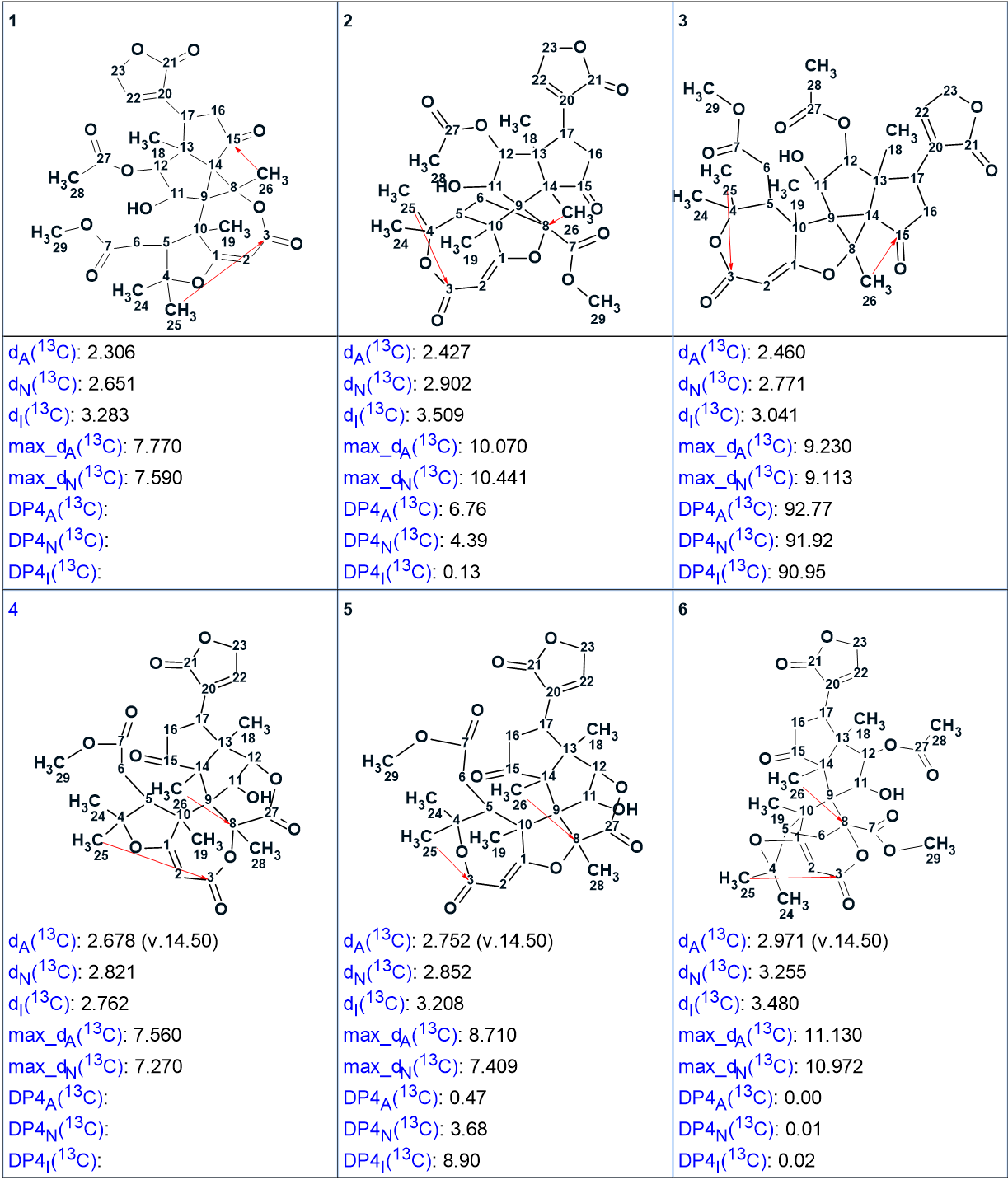
Figure 3. DP4 probabilities calculated for valid possible structures.
It is clearly visible that structure #3 has the highest DP4 probability. The application of CASE allows one to determine all structures satisfying the 1D and 2D NMR data and then select the most probable structure in the output file [3]. As we see, the implementation of the DP4 methodology into the ACD/SE expert system made it possible to define the probability of the best structure numerically, in this case.
Therefore, ACD/Structure Elucidator confirmed the structure of trichilianone D as suggested in the article [1]:
Note that the solution to the problem was found in a fully automatic mode, and the right structure was selected by using DP4 style metrics. More accurate results can be obtained if DFT calculations are used to predict the chemical shifts and coupling constants of the structures, as described previously [4-6].
References
- Limachi, I., Gonzalez-Ramirez, M., Manner, S., Ticona, J.C., Salamanca, E., Gimenez, A., and Sterner, O. (2021). Trichilianones A-D, Novel Cyclopropane-Type Limonoids from Trichilia adolfi. Molecules, 26, 1019. 10.3390/molecules26041019
- Ermanis, K., Parkes, K.E.B., Agback, T., Goodman, J.M. (2017). Doubling the power of DP4 for computational structure elucidation. Org. Biomol. Chem., 15(42), 8998–9007. 10.1039/C7OB01379E
- Elyashberg, M.E.; Williams, A.J.; Blinov, K.A. Contemporary Computer-Assisted Approaches to Molecular Structure Elucidation; RSC, Cambridge, 2012. http://pubs.rsc.org/en/Content/eBook/978-1-84973-432-5
- Buevich, A.V., Elyashberg, M.E. (2016). Synergistic combination of CASE algorithms and DFT chemical shift predictions: a powerful approach for structure elucidation, verification and revision. J. Nat. Prod., 79(12), 3105–3116. 10.1021/acs.jnatprod.6b00799
- Buevich, A.V., Elyashberg, M.E. (2018). Towards unbiased and more versatile NMR-based structure elucidation: A powerful combination of CASE algorithms and DFT calculations. Magn. Reson. Chem., 56, 493–504. 10.1002/mrc.4645
- Buevich, A.V., Elyashberg, M.E. (2020). Enhancing Computer Assisted Structure Elucidation with DFT analysis of J-couplings. Magn. Reson. Chem., 58(6), 594–606. 10.1002/mrc.4996
About the Author
