July 1, 2014
by Mikhail Elyashberg, Leading Researcher, ACD/Labs
Trigoflavidol A
Tang et al [1] carried out a phytochemical investigation on the stems of T. flavidus collected in China. They reported the identification of five degraded diterpenoids, including the tetranorditerpenoid dimers trigoflavidols A and B and the hexanorditerpenoid Trigoflavidol C. Spectroscopic data acquired for Trigoflavidol A (1), possessing a rearrangement skeleton with a spiroketal core moiety, was used for challenging ACD/Structure Elucidator Suite. In this example we present three different methods to solve the correct structure.
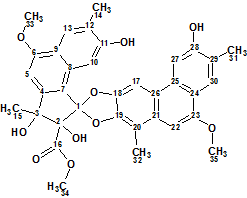
1
Trigoflavidol A was determined to have the molecular formula C35H32O10 with 20 degrees of unsaturation based on the [M + Na]+ ion at m/z 635.1898 (calculated 635.1893) in its positive HR-ESI-MS. Considering the high degree of unsaturation (a significant deficit of hydrogen atoms) one can expect that the unknown will be difficult to identify in the “ab initio” mode due to a potential lack of 2D NMR correlations.
A strong absorption band in the IR spectrum at 3430 cm-1 bears witness to the presence of hydroxyl groups. The IR absorptions at 1631, 1588, and 1469 cm-1 suggest the presence of phenyl functionalities. A band observed at 1728 cm-1 hints at the presence of ester group(s). The modest intensity of the band can be explained by the “low concentration” of C=O groups in a fairly large molecule (m/z =635 au).
The authors [1] give the following structural interpretation of the NMR data. The 35 carbon signals observed in the 13C NMR and DEPT spectra were classified as seven methyl groups including three O-methyl groups, seven methines, and 21 quaternary carbons including one ester carbonyl, six oxygenated aromatic carbons, and three other oxygenated carbons. In addition, one tertiary methyl at δH 1.61 (3H, s), three aromatic methyls at δH 2.33, 2.42, and 2.45 (each 3H, s), seven uncoupled aromatic protons at δdelta;H 6.90, 7.00, 7.20, 7.62, 7.90 (each 1H, s), and 8.05 (2H, s), and four hydroxy protons at δH 4.71, 4.77 (each 1H, s) and 8.89 (2H, s) were distinguished through further analysis of the NMR spectra. The comparison of the aforementioned values with the spectroscopic data related to a natural product Actephilol A (2) [2] led the authors to conclude that Trigoflavidol A was a heterodimer comprised of two different highly aromatized tetranorditerpenoids.
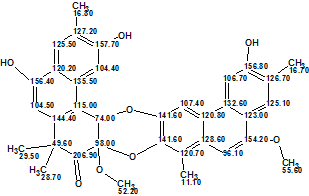
2
Thus, the structure of Trigoflavidol A was derived by Tang et al on the basis of jointly employing experimental spectroscopic data and the spectroscopic data of Actephilol A. We made an attempt to elucidate the structure of unknown using only the tabulated 1D NMR and HSQC data in combination with HMBC correlations graphically presented in the work [1]. The input data are presented in Table 1.
Table 1: Trigoflavidol A. Spectroscopic NMR data
| Label | δX | δC cld | XHn | δH | M(J) | HMBC |
| C 1 | 121.9 | 119.49 | C | |||
| C 2 | 92.9 | 90.8 | C | |||
| C 3 | 80.9 | 83.33 | C | |||
| C 4 | 149.8 | 153.16 | C | |||
| C 5 | 96.4 | 104.72 | CH | 6.9 | u | C 9, C 7, C 3 |
| C 6 | 159.6 | 159.05 | C | |||
| C 7 | 121.5 | 119.24 | C | |||
| C 8 | 131.4 | 128.13 | C | |||
| C 9 | 119.6 | 127.85 | C | |||
| C 10 | 106.26 | 110.72 | CH | 7.2 | u | C 9, C 7, C 12 |
| C 11 | 156.8 | 155.37 | C | |||
| C 12 | 127.1 | 125.49 | C | |||
| C 13 | 125.2 | 123.45 | CH | 8.06 | u | C 6, C 11, C 8 |
| C 14 | 17.1 | 15.98 | CH3 | 2.33 | u | C 13, C 12, C 11 |
| C 15 | 27.8 | 23.28 | CH3 | 1.61 | u | C 4, C 2 |
| C 16 | 173.3 | 168.79 | C | |||
| C 17 | 98.4 | 103.24 | CH | 7.62 | u | C 19, C 25, C 21 |
| C 18 | 147.09 | 144.39 | C | |||
| C 19 | 147.12 | 143.8 | C | |||
| C 20 | 112.3 | 119.87 | C | |||
| C 21 | 128.9 | 127.49 | C | |||
| C 22 | 96.8 | 95.21 | CH | 7 | u | C 24, C 20, C 26 |
| C 23 | 153.5 | 149.69 | C | |||
| C 24 | 119.9 | 118.29 | C | |||
| C 25 | 132.6 | 129.21 | C | |||
| C 26 | 121.9 | 119.02 | C | |||
| C 27 | 106.33 | 105.63 | CH | 7.9 | u | C 29, C 24, C 26 |
| C 28 | 156.4 | 154.64 | C | |||
| C 29 | 126.1 | 125.7 | C | |||
| C 30 | 125 | 123.65 | CH | 8.05 | u | C 28, C 31, C 25, C 23 |
| C 31 | 16.8 | 16.5 | CH3 | 2.42 | u | C 30, C 29, C 28 |
| C 32 | 11.7 | 9.82 | CH3 | 2.45 | u | C 19, C 21 |
| C 33 | 56.2 | 55.76 | CH3 | 4.09 | u | C 6 |
| C 34 | 53.1 | 52.27 | CH3 | 3.62 | u | C 16 |
| C 35 | 55.7 | 55.13 | CH3 | 4.1 | u | C 23 |
| O 1 | 100* | OH | 4.77 | u | C 3, C 2, C 16, C 1 | |
| O 2 | 110* | OH | 4.71 | u | C 15, C 2, C 4, C 3 | |
| O 3 | 120* | OH | 8.89 | u | C 12, C 11, C 10 | |
| O 4 | 130* | OH | 8.9 | u | C 27, C 29, C 28 |
* Fictitious 17O chemical shifts.
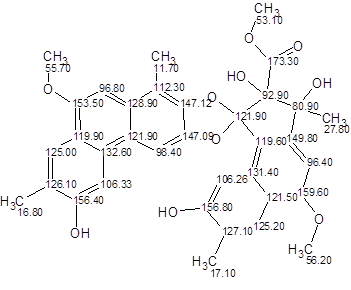
The Molecular Connectivity Diagram (MCD) created by ACD/Structure Elucidator Suite based on this information is shown in Figure 1.
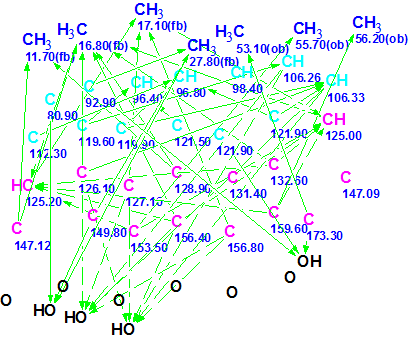
Figure 1: Trigoflavidol A Initial Molecular Connectivity Diagram
Method 1. In the chemical shift range of 80-122 ppm, the MCD contains 13 carbon atoms (marked in blue) which can be assigned either as sp3/2ob (obligatory) in the acetal substructure or as sp2/fb (forbidden). There are 15 13C signals in the narrow range of 100-130 ppm, i.e. the 13C NMR spectrum is fairly congested in this region. Some carbons emerge with only one connectivity, while carbon C(147.09) has no connectivities at all. These observations led to the conclusion that available structural constraints produced from NMR data are scarce enough. Taking into account characteristic 1H chemical shifts, the hybridization of the carbon atoms in the range 96-106 ppm can be set as sp2, while atoms C 80.9 and C 92.9 – as oxygenated carbons sp3. Carbons C 159.6 and C 173.3 are likely connected to oxygen atoms and they were supplied with a label “ob“. Based on accumulated experience gathered while solving CASE problems it was predicted that the time for structure generation, even from an edited MCD, would be extremely long. Nevertheless, for the sake of computational experiment completeness, structure generation combined with 13C chemical shift prediction (options d<4 and d(max)<20 ppm) was initiated. It was interrupted after many hours when ~1,800,000 structures were generated but no structure was stored: all generated structures were rejected by filter. It became evident that additional structural constraints were crucially necessary to find a solution to the problem. In such a situation it is desirable to try to introduce some fragments capable of “absorbing” as many atoms displayed on the MCD as possible. Because the presence of phenyl substructures is evident, an attempt was first made to recognize on the MCD those carbon atoms that can be involved in forming the benzene rings (see our previous Elucidation of the Month problem regarding Spiroindimicin A). Two benzene rings were distinguished and the corresponding atoms were connected with connectivities of one bond length (Figure 2).
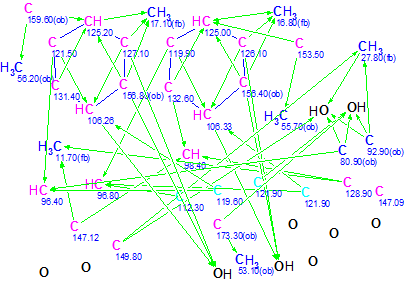
Figure 2: Trigoflavidol A Molecular Connectivity Diagram with two benzene rings selected manually.
Structure generation from the modified MCD gave the following result: k = 1458 → 1, tg = 11 s, dA=2.68, dN= 2.84, dI =3.18, with the unique structure 3 coinciding with Trigoflavidol A.
3
The problem was solved due to the successful selection of two benzene rings in the MCD. However application of this approach implies some user skill and experience. It was interesting to try another method of problem solving that
would be more unbiased.
Method 2. An attempt was made to solve the problem using a fragment search by 13C NMR spectrum in the ACD/Labs Fragment Library. As a result of the search, 1103 fragments were found (L=1103). To select the most appropriate fragments for creating MCDs it is desirable to reveal such fragments which would be large enough to absorb as many as possible free atoms and at the same time obtain those 13C sub-spectra that would be as close as possible to the experimental one. For this purpose the first 35 fragments ranked in descending order of carbon atom numbers were selected (l=35) and this subset of Found Fragments was ranked in ascending order of sub-spectrum deviations dE from the corresponding experimental 13C sub-spectrum. The top fragments of the ranked file are displayed in Figure 3.
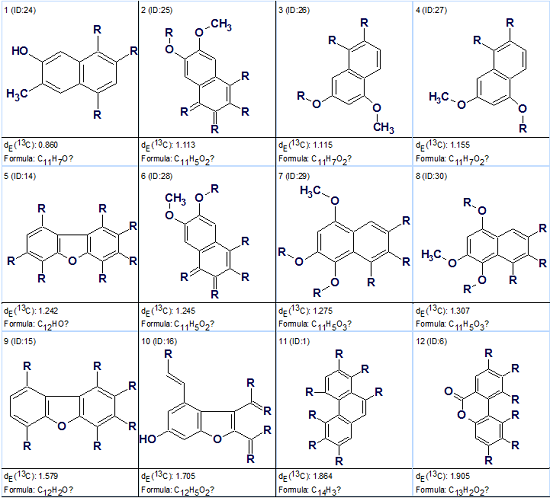
Figure 3: Trigoflavidol A. Top structures of the Found Fragments ranked file
Figure 3 shows that the first ranked fragment ID:24 has minimum deviation and its sub-molecular formula is C11H7O, so the fragment encompasses 12 of 45 skeleton atoms (more than 25%). Figure 4 shows the structure of the fragment ID:24 along with its 13C sub-spectrum (upper) and a corresponding part of the experimental spectrum of unknown (lower).Visual comparison of the spectra leads to the conclusion that the sub-spectrum of the fragment fits rather well with the experimental spectrum.
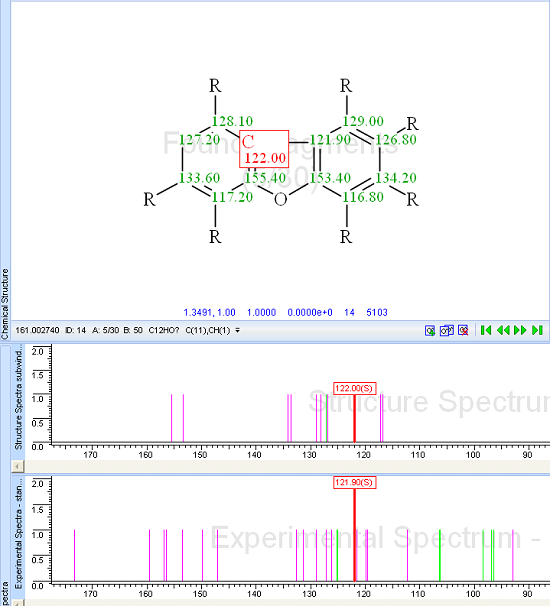
Figure 4: Trigoflavidol A. Structure of the fragment ID:24 along with its 13C subspectrum (upper) and a corresponding part of the experimental spectrum of compound (lower).
The fragment ID:24 was displayed in the Found Fragments window, and a command “Create MCDs for SBG from current structure” was activated. By gradually increasing the tolerance T for fitting the sub-spectrum to the experimental spectrum, the value T=6.5 ppm was reached, at which point 262 MCDs were created by the program. The first 189 MCDs contained two fragments ID:24, while the remaining MCDs contained only one fragment with different carbon atom assignments.
Structure generation from 262 MCDs combined with 13C chemical shift prediction and filtering gave the following result: k = 4,149,035→4→1, tg= 20 h, and the single output structure again coincided with structure of Trigoflavidol A. The problem-solving protocol showed that the first 189 double-fragmented MCDs were immediately rejected during the structure generation. More than four million structures were generated from the remaining MCDs.
Thus, application of the Found Fragments allowed us to avoid manual selection of two benzene rings on MCD and an unambiguous solution was found almost automatically. The “price of victory” was a long time for structure generation.
We considered two different approaches to solve the problem and they both assumed that the researcher uses only information extracted from spectra and the system knowledge. But in reality the researcher frequently knows a set of earlier identified structures belonging to the chemical family to which the unknown can be related. For instance, a series of similar compounds are usually isolated from a given natural object. Particularly, to elucidate the structure of Trigoflavidol A, the authors[1] used the structure and assigned 13C and 1H NMR spectra of Actephilol A, 2 [2]. We will show how this a priori information can simplify the elucidation of the structure of Trigoflavidol A by making it automatic.
Method 3. Structure Elucidator Suite provides the possibility to create a User Database (UDB). In this problem the structure of Actephilol A was used to produce the UDB. For this goal, a standard procedure was applied to structure 2, and as a result a UDB containing 96 fragments was created. A fragment search by 13C NMR spectrum in the UDB was completed with the selection of 33 fragments to which the command “Create MCD(s) from Found Fragments” was applied. The program produced 72 MCDs, and one of them is presented in Figure 5.
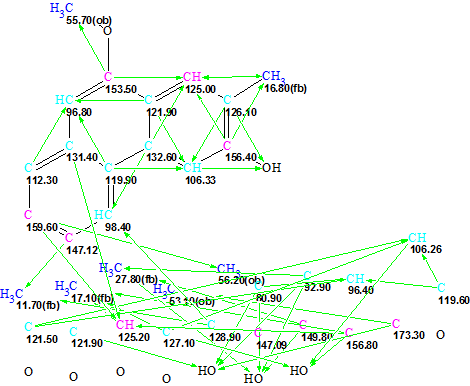
Figure 5: Trigoflavidol A. An example of the MCD created from the Found Fragments that were selected as a result of a fragment search in the User Database
Structure generation accompanied by 13C chemical shift prediction gave the following result: k=232464→4→1, tg = 15 h and the single structure again coincided with structure 1. The time of structure generation was reduced by 5 h but still remained quite long. However, this was under a condition where no user assumptions were used except for the assumption that Actephilol A and the unknown compound belong to the same family of natural products.
References
1. Tang GH, Zhang Y, Gu YC, Li SF, Di YT, Wang YH, Yang CX, Zuo GY, Li SL, He HP, Hao XJ (2012) Trigoflavidols A-C, degraded diterpenoids with antimicrobial activity, from Trigonostemon flavidus. J Nat Prod 75 (5):996-1000. doi:10.1021/np3001128
2. Ovenden SPB, Yew ALS, Glover RP, Ng S, Rossant CJ, Regalado JC, Soejarto DD, Buss AD, Butler MS (2001) Actephilol A and epiactephilol A: two novel aromatic terpenoids isolated from Actephila excelsa. Tetrahedron Lett 42:7695-7697
About the Author
