November 1, 2017
by Mikhail Elyashberg, Leading Researcher, ACD/Labs
Uncarilin A
It is known that melatonin (5-methoxy-N-acetyltryptamine, MT), an endogenous hormone, plays an important role in regulating circadian rhythms. This happens by activating two G-protein-coupled receptors, MT1 and MT2. These are good targets for drug discovery for drugs treating insomnia and sleep disorders.
The traditional Chinese medicine Gou-Teng is used for treating neurological disorders. Its main chemical constituents are indole alkaloids, flavonoids, triterpenoids, and organic acids. The indole alkaloids are characteristic constituents responsible for the hypotensive action of Gou-Teng. Five Uncaria plant species are documented as the sources of Gou-Teng according to the latest Chinese Pharmacopoeia (2015).
In the search for novel MT receptor agonists from natural sources, Geng at al [1] used LC-MS to study U. rhynchophylla,, resulting in the isolation of two pairs of dimeric isoechinulin-type enantiomers, (±)-uncarilin A (1) and (±)-uncarilin B:
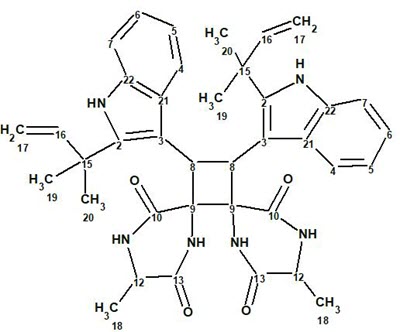
1
Spectroscopic data used for elucidating the structure of compound 1 were utilized to challenge ACD/Structure Elucidator. The positive HR-MS spectrum displayed a protonated molecular ion at m/z 647.3341 ([M + H]+, +0.1 mDa), corresponding to a molecular formula of C38H42N6O4, indicating 21 degrees of unsaturation. The UV absorptions at 283 and 226 nm point to the presence of an indole chromophore in the structure. A strong IR absorption band at 1660 cm-1 shows the presence of an amide group. 1H, 13C, and HSQC data were fully tabulated in the article [1], while only key COSY and HMBC correlations were presented in a graphical form on the elucidated structure 1. The molecular formula of this compound contains 38 carbon atoms, while only 19 signals were observed in the 13C NMR. This is a very clear indication that the molecule under investigation is symmetric. The available NMR spectroscopic data are presented in Table 1.
Table 1. Spectroscopic NMR data
| C/X Label | δC | δC calc* | XHn | δH | M(J) | COSY | H to C HMBC |
|---|---|---|---|---|---|---|---|
| C 2 | 144.9 | 141.54 | C | ||||
| C 3 | 106.7 | 106.13 | C | ||||
| C 4 | 122.6 | 118.87 | CH | 9.38 | d(8.1) | 7.43 | C 3, C 22 |
| C 5 | 120.1 | 118.77 | CH | 7.43 | t(8.1) | 7.20, 9.38 | |
| C 6 | 121.2 | 121.62 | CH | 7.2 | t(8.1) | 7.38, 7.43 | |
| C 7 | 111.7 | 110.61 | CH | 7.38 | d(8.1) | 7.2 | C 21 |
| C 8 | 46.2 | 42.69 | CH | 6.38 | s | C 2, C 10 | |
| C 9 | 67.3 | 66.22 | C | ||||
| C 10 | 171 | 166.36 | C | ||||
| C 12 | 49.9 | 49.53 | CH | 4.15 | q(7.0) | 1.45 | |
| C 13 | 168 | 169.32 | C | ||||
| C 15 | 39.2 | 38.98 | C | ||||
| C 16 | 145.9 | 146.72 | CH | 6.37 | dd(17.3,10.5) | 5.3 | C 2 |
| C 17 | 112.9 | 111.63 | CH2 | 5.12 | d(10.5) | ||
| C 17 | 112.9 | 111.63 | CH2 | 5.3 | d(17.3) | 6.37 | C 15 |
| C 18 | 18.6 | 19.77 | CH3 | 1.45 | d(7.0) | 4.15 | C 13 |
| C 19 | 29.2 | 28.01 | CH3 | 1.56 | s | C 2, C 16 | |
| C 20 | 28.5 | 28.01 | CH3 | 1.33 | s | C 2, C 16 | |
| C 21 | 129.3 | 127.46 | C | ||||
| C 22 | 136.2 | 136.17 | C | ||||
| N 1 | NH | 11.51 | s | C 3, C 21 | |||
| N 2 | NH | 10.16 | u | C 9, C 12, C 13 | |||
| N 3 | NH | 10.52 | s | C 9, C 10, C 12 |
* 13C chemical shift calculations were carried out using a HOSE code based approach [2]
The Molecular Connectivity Diagram (MCD) automatically generated by these data is presented in Figure 1.
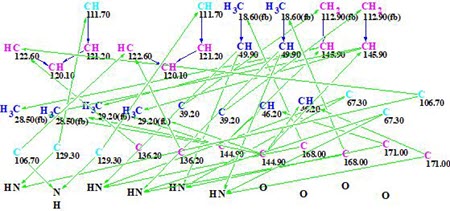
Figure 1. Initial Molecular Connectivity Diagram (MCD) automatically generated by the program.
MCD overview. The MCD shows that four (eight) carbon atoms C 111.7, C 129.3, C 106.7, and C 63.7 are colored light blue, which means that hybridizations of these atoms are sp2 or sp3 (not sp). As the IR spectrum indicates, the presence of a NH-C=O group, four carbonyl bonds were drawn manually at carbons C 168.0 and C 171. Taking this into account, hybridizations and properties sp2/ fb (forbidden to bond with a heteroatom) were set for atoms C 111.7, C 129.3, and C 106.7, while sp3/ob (obligatory to bond with a heteroatom) for atom C 63.7. The hybridizations and properties (ob or fb) of other carbon atoms were set by the program automatically. 1H signal multiplicities displayed in Table 1 (column M(J)) were used to set the numbers of hydrogen atoms attached to carbons existing in the first sphere environment of a given CHn group. The edited MCD is shown in Figure 2.
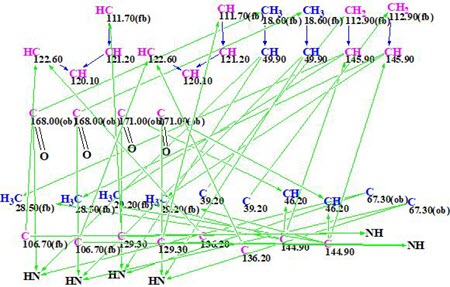
Figure 2. Edited molecular connectivity diagram.
Strict structure generation accompanied with 13C chemical shift prediction (incremental approach was used in this stage) and structure filtering was initiated from the edited MCD, which returned the following results: k =117064 → 71 → 59, tg = 17 min. Then, as usual, further 13C chemical shift predictions by HOSE code based method (average deviations are denoted as dA) and by neural networks algorithm (average deviations are denoted as dN) were performed, and the output file was ranked in increasing order of dA values. The six top ranked structures are presented in Figure 3.
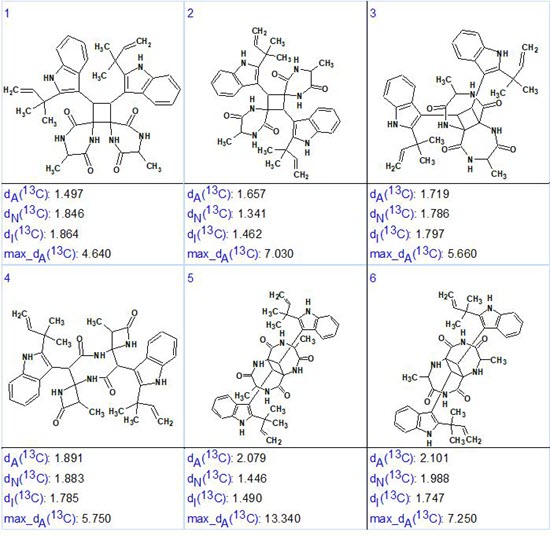
Figure 3. The six top ranked structures of the output file.
We see that the structure which is identical to uncarilin A (1) is ranked first but the difference between the average deviations calculated for the first two structures (#1 and #2) is small. Additionally, values dN and dI favor structure #2! Also maximum deviation max_dA is minimal for structure #1. These discrepancies are caused by the high similarity of structures #1 and #2. In such situations, additional confirmation of the most probable structure is necessary. An obvious resolution would be to record NOESY or ROESY spectra for the compound. In these, an interaction between the protons of atoms 17, 19 and 20 (5.3, 1.56, and 1.33 ppm) with the aromatic protons on atoms 4, 5 and 6 (9.38, 7.43, and 7.2 ppm) should be visible but probably weak, indicating the validity of structure #1. Such correlations will not be visible for structure #2. Unfortunately the ROESY spectrum in the Supporting Information of [1] is not that informative for this as it only displays the strongest correlations. Alternatively NMR chemical shift prediction by the DFT approach can be used [3,4] or X-ray crystallography. In the article [1] structure #1 was proved by X-ray diffraction and ECD spectroscopic data.
The final structure #1 with 13C chemical shift assignments carried out by ACD/Structure Elucidator is shown below:
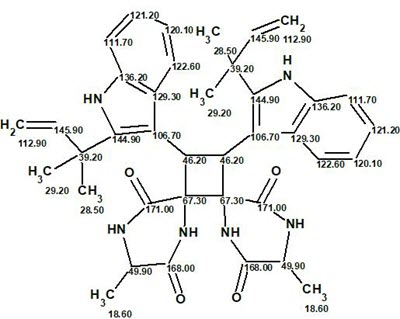
In conclusion, another complex symmetric molecule was elucidated by ACD/Structure Elucidator with minimum user intervention in a relatively small
amount of time.
References
- C.-A. Geng, X.-Y. Huang, Y.-B. Ma, B. Hou, T.-Z. Li, X.-M. Zhang, J.-J. Chen. (2017). (±)-Uncarilins A and B, Dimeric Isoechinulin-Type Alkaloids from Uncaria rhynchophylla. J. Nat. Prod., 80: 959-964.
- M.E. Elyashberg, A.J. Williams. (2015). Computer-based Structure Elucidation from Spectral Data (p. 454). Springer-Verlag Berlin, Heidelberg.
- A. V. Buevich, M. E. Elyashberg. (2016). Synergistic combination of CASE algorithms and DFT chemical shift predictions: a powerful approach for structure elucidation, verification and revision. J. Nat. Prod., 79 (12): 3105–3116.
- A.V. Buevich, M. E. Elyashberg. (2017). Towards unbiased and more versatile NMR-based structure elucidation: A powerful combination of CASE algorithms and DFT calculations. Magn. Reson. Chem., DOI: 10.1002/mrc.4645
About the Author
