November 1, 2018
by Mikhail Elyashberg, Leading Researcher, ACD/Labs
Actinobenzoquinoline
In the quest for new microbial strains that could produce novel natural products the exploration of diverse and poorly examined environments is important. The group of W. Fenical in UCSD has been looking into marine sendiments as sources of actinomycete bacteria that would be different from terrestrial strains. In this process the strain CNQ-149 was isolated from such a marine sediment, which was collected close to San Diego, CA, USA. Several unknown compounds with molecular weights between 300-400 Da and long UV wavelength chromophores were identified after studies with LC-MS of the crude extract. In order to isolate some of these a large scale fermentation was performed, followed by repeated purification of the organic extract. This led to the isolation of four new alkaloids: actinobenzoquinoline (1) and actinophenanthrolines A−C, belonging to two unprecedented natural alkaloid classes. This process is described in [1], together with the structure elucidation of these four alkaloids. The suggested structure 1 with its stereochemistry was confirmed by X-ray diffraction.
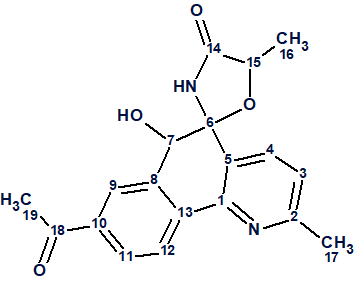
1
The spectroscopic data presented for this structure in article [1] were used to challenge ACD/Structure Elucidator.
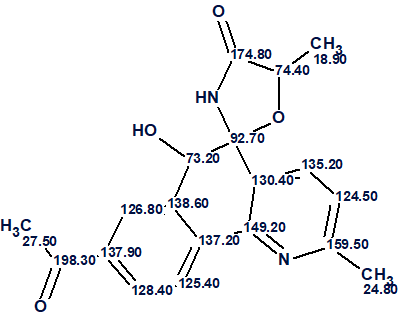
Actinobenzoquinoline (1) was isolated as a pale yellow solid, with a molecular formula of C19H18N2O4 based on a sodium adduct ion at m/z 361.1153 [M + Na]+ in the HR-ESI-MS data, together with 13C NMR data. The article only listed 1H–1H COSY and key 1H–13C HMBC correlations (Figure 1).
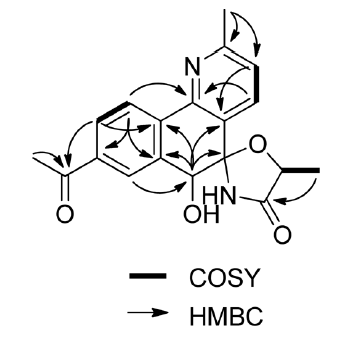
Figure 1. COSY and key HMBC correlations used in the structure elucidation of actinobenzoquinoline [1].
The spectroscopic data entered into ACD/Structure Elucidator are presented in Table 1.
Table 1. 1D and 2D NMR spectroscopic data.
| Label | C | C calc* | XHn | H | M | COSY | H to C HMBC |
| C 1 | 149.200 | 156.250 | C | ||||
| C 2 | 159.500 | 158.500 | C | ||||
| C 3 | 124.500 | 122.540 | CH | 7.360 | d | 7.87 | C 5 |
| C 4 | 135.200 | 135.390 | CH | 7.870 | d | 7.36 | C 6, C 1, C 2 |
| C 5 | 130.400 | 128.850 | C | ||||
| C 6 | 92.700 | 87.760 | C | ||||
| C 7 | 73.200 | 65.870 | CH | 4.900 | s | C 6, C 9, C 5, C 13, C 8 |
|
| C 8 | 138.600 | 136.390 | C | ||||
| C 9 | 126.800 | 127.990 | CH | 8.120 | u | C 7, C 11, C 13, C 18 |
|
| C 10 | 137.900 | 135.530 | C | ||||
| C 11 | 128.400 | 128.610 | CH | 8.010 | d | 8.31 | C 9, C 13, C 18 |
| C 12 | 125.400 | 128.990 | CH | 8.310 | d | 8.01 | C 10, C 8, C 1 |
| C 13 | 137.200 | 136.750 | C | ||||
| C 14 | 174.800 | 172.810 | C | ||||
| C 15 | 74.400 | 75.360 | CH | 4.610 | q | 1.34 | C 14 |
| C 16 | 18.900 | 17.830 | CH3 | 1.340 | d | 4.61 | C 15, C 14 |
| C 17 | 24.800 | 24.840 | CH3 | 2.570 | s | C 3, C 2 |
|
| C 18 | 198.300 | 198.010 | C | ||||
| C 19 | 27.500 | 26.460 | CH3 | 2.620 | s | C 18 | |
| N 1 | NH | 9.100 | u | ||||
| O 1 | OH | 6.270 | u |
* 13C chemical shift calculations were performed using the HOSE code based approach.
Figure 2 shows the Molecular Connectivity Diagram (MCD) which was created automatically by the program and slightly edited by the user.
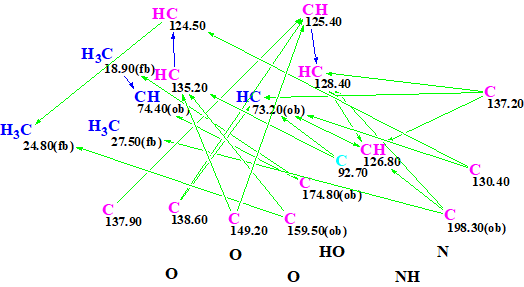
Figure 2. Molecular Connectivity Diagram.
As all 2D NMR connectivities are of a standard length (Figure 1) a strict structure generation was initiated, which gave the following results: k = 838 → 333 → 333, tg = 34 m.
Afterwards 13C chemical shift prediction was performed for all stored structures using three empirical methods intrinsic for ACD/Structure Elucidator (neural networks, incremental and HOSE code based), and the structural output file was ranked in increasing order of averaged deviations dA(13C) calculated by HOSE code based approach. The first eight structures of the ranked output file are presented in Figure 3.
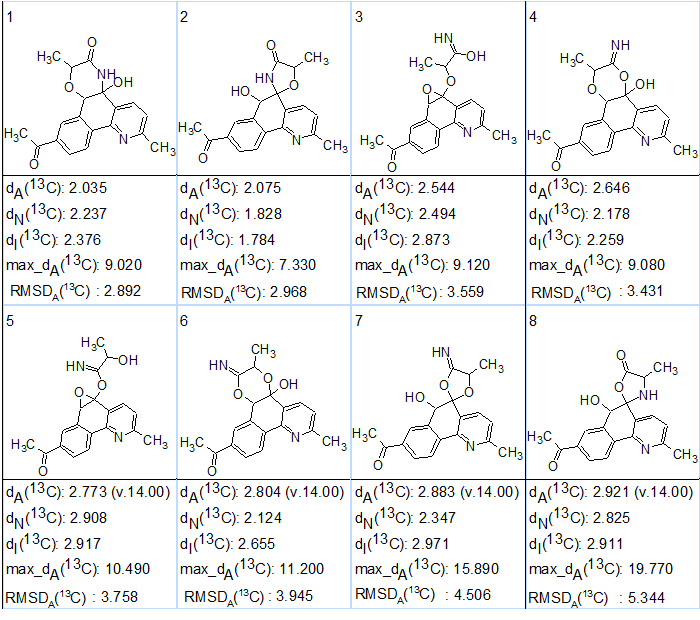
Figure 3. The eight highest ranked structures suggested by the CASE program for actinobenzoquinoline.
We see that the structure of actinobenzoquinoline determined in [1] has been generated by the program and ranked in the second position of the structural file (#2). In the same time, the dA(13C) deviations found for the two first structures are almost equal and hence a reliable selection of a right structure is impossible with the given data. As has been shown [2], in such a situation, and in order to resolved this ambiguity, the structure ranking can be checked by NMR chemical shift prediction on the basis of DFT calculations. The corresponding calculations were carried out in [2] in accordance with methodology suggested in [3].
The DFT analysis of the carbon chemical shifts of the first six structures predicted by CASE analysis was done for all four possible stereoisomers of structures #1, #2, #4, and #6 and two possible stereoisomers of structures #3 and #5 (see Figure 4).
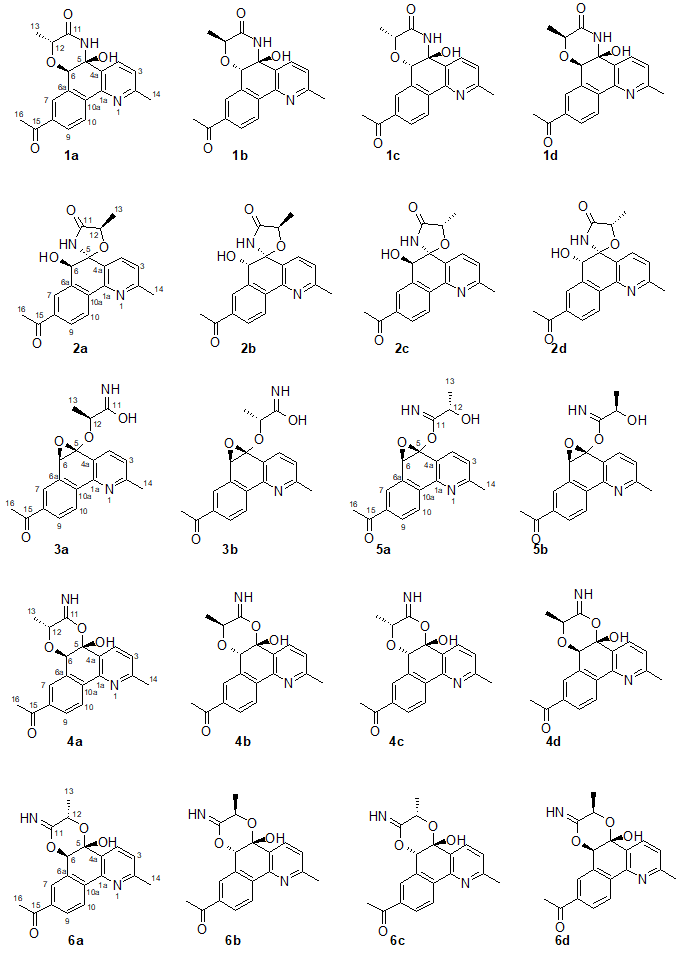
Figure 4. Six top-ranked candidates structures and their stereoisomers (labeled with a, b, c, and d indices) for actinobenzoquinoline.
DFT calculations of the carbon chemical shifts were done at the mPW1PW91/6-311+G(2d,p) level of theory with inclusion of the polarizable continuum model for DMSO (scrf = (solvent =dimethylsulfoxide)). Carbon chemical shift values were then derived from calculated isotropic shielding constants by applying the following scaling coefficients: 186.2534 (intercept) and −1.0496 (slope). Prior to DFT chemical shift calculations molecular geometries were optimized at the B3LYP/6-31+G(d,p) level of DFT. Only the lowest energy conformation for each isomer were considered in this analysis. As shown in Figure 5, the lowest RMSD and max_d parameters were indeed found for stereoisomers of structure #2, which allowed to confirm structure 1.
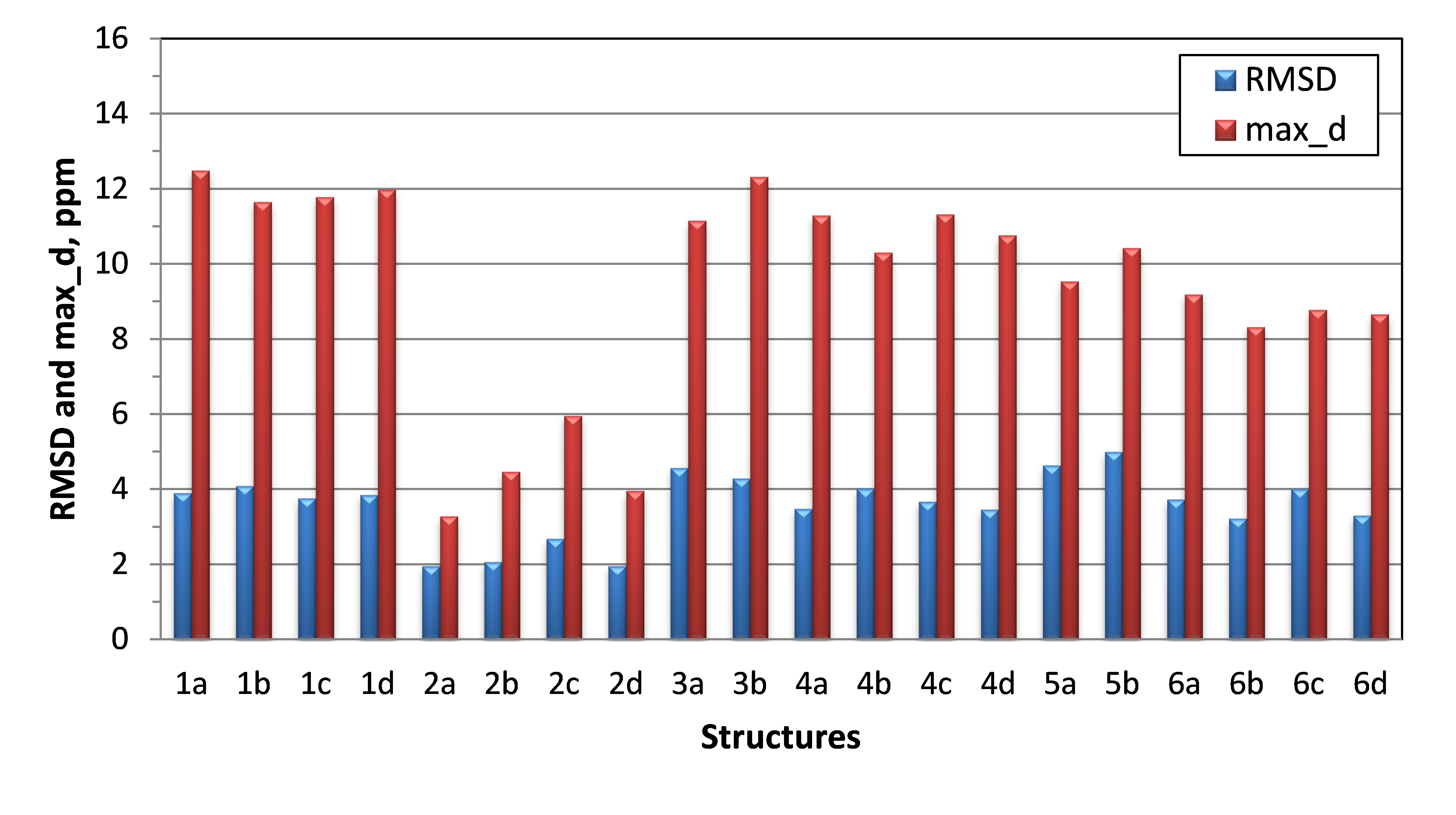
Figure 5. RMSD and max_d between experimental and DFT-calculated δ(13C) for six top-candidate structures of actinobenzoquinoline and their stereoisomers a, b, c, and d. Structures from this figure are referred to in the text as 1a, etc.
Next an attempt was made [2] to reconfirm the correct stereoisomer actinobenzoquinoline 2d (it has been previously determined by X-ray crystallography in [1]) by using DFT chemical shift analysis. As can be seen in Figure 5, when the analysis of chemical shifts was done only for the lowest energy rotamer, the correct stereoisomer 2d had the same RMSD as 2a and 2b. This is a rather challenging problem requiring more rigorous chemical shift analysis for a flexible molecule such as actinobenzoquinoline. The problem was further exacerbated by the fact that experimental NMR data were collected in DMSO solution which is more difficult to reproduce by DFT calculations compared with other solvents like chloroform.
2d
A full conformational analysis of each of the four isomers of structure #2 (see Figure 6) was performed using the previously described method based on comprehensive conformational analysis by force field calculation followed by cluster analysis and DFT optimization of the lowest energy conformations for each cluster [4].
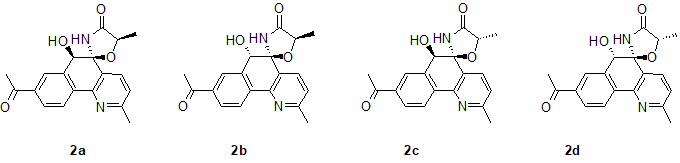
Figure 6. The four possible stereoisomers of actinobenzoquinoline . Structure numbers in this figure refer to the CASE output structures shown in Figure 6.
This analysis generated sets of 7, 7, 10 and 6 conformations for 2a, 2b, 2c and 2d stereoisomers (Figure 6), respectively. Note that stereoisomer 2d is consistent with the structure of actinobenzoquinoline [1]. For geometry and energy optimization three different DFT levels of theory were tested, which included hybrid functional B3LYP and dispersion-corrected functionals B3LYP-D2 and M06-2X. Geometry optimizations were done with the 6-31+G(d,p) basis set and with inclusion of the polarizable continuum model for DMSO (scrf = (solvent=dimethylsulfoxide)). For each conformational ensemble two sets of NMR chemical shifts were calculated for chloroform and DMSO as solvents approximated with the polarizable continuum model (PCM) at the mPW1PW91/6-311+G(2d,p) level of theory.
Conformationally averaged chemical shifts for four isomers of structure #2 allowed to select the best stereoisomer. The lowest 13C chemical shift RMSD (1.64 ppm) was indeed found for the correct stereoisomer 2d (mPW1PW91/6-311+G(2d,p)// B3LYP/6-31+G(d,p)). Dispersion corrected functionals B3LYP-D2 and M06-2X, on average, produced lower accuracy data and could not differentiate the right stereoisomer.
As a result utilization of the methodology described in [2,3] allowed for selecting the correct structure of actinobenzoquinoline among the top ranked structures suggested by ACD/Structure Elucidator:
The difficult problem of analysis of the conformationally flexible, chiral molecule of actinobenzoquinoline was successfully solved and the relative stereochemistry of this molecule was determined on the basis of DFT calculations without the need to us X-ray diffractions methods as the authors of [1] did.
References
- S.-J. Nam, C. A. Kauffman, P. R. Jensen, C.E. Moore, A.L. Rheingold, W. Fenical. (2015). Actinobenzoquinoline and Actinophenanthrolines A−C, Unprecedented Alkaloids from a Marine Actinobacterium. Org. Lett., 17: 3240−3243.
- A.V. Buevich, M. E. Elyashberg. (2018). Towards unbiased and more versatile NMR-based structure elucidation: A powerful combination of CASE algorithms and DFT calculations. Magn. Reson. Chem., 56: 493–504.
- A. V. Buevich, M. E. Elyashberg. (2016). Synergistic combination of CASE algorithms and DFT chemical shift predictions: a powerful approach for structure elucidation, verification and revision. J. Nat. Prod., 79 (12): 3105–3116.
- E. C. Sherer, C. H. Lee, J. Shpungin, J. F. Cuff, C. Da, R. Ball, R. Bach, A. Crespo, X. Gong, C. J. Welch. (2014). Systematic Approach to Conformational Sampling for Assigning Absolute Configuration Using Vibrational Circular Dichroism. J. Med. Chem., 57(2): 477−494. DOI: 10.1021/jm401600u
About the Author
