January 1, 2022
by Mikhail Elyashberg, Leading Researcher, ACD/Labs
Asperspiropene A
Asperspiropene A (1) was first reported by He et al [1] as the second derivative of a class of compounds containing a unique six-membered ring substructure. The molecular formula was determined from the HR-MS spectrum to be C17H26O8.
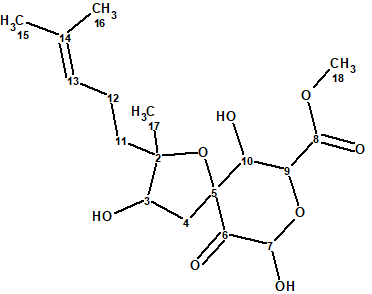
1
Asperspiropene A showed a potent inhibitory effect on the cancer-associated mutant isocitrate dehydrogenase and its structure−activity relationship was investigated by a molecular docking study.
Cao et al [2] have isolated a number of oxygenated cyclohexenone derivatives from two strains of Aspergillus flocculosus, 01NT-1.1.5 and 168ST-16.1. Among them there was a compound with identical 1D and 2D NMR data to those of asperspiropene A. The HR-ESI-MS data of this, however, were different from the previously published data for asperspiropene A. Additionally, it was found that some 2D NMR data of asperspiropene A (COSY and HMBC) were incorrectly interpreted. These anomalies prompted Cao et al [2] to carefully analyze the spectroscopic data of 1 and to revise the structure of asperspiropene A as sown in Figure 1.
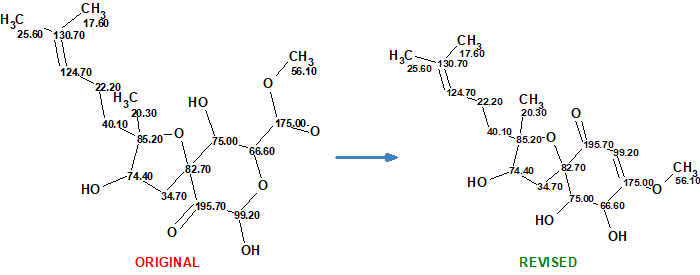
Figure 1. Original and revised structures of asperspiropene A.
We used the 1D and 2D NMR data published in the original article [1] to demonstrate how the revision could be accomplished if ACD/Structure Elucidator (ACD/SE) is used. First, the originally published structure was defined as a Proposed Structure (PS) in the program and 13C chemical shift prediction was performed, in order to see how well the experimental data matched with the predicted ones. This was done using the three algorithms included with ACD/SE: incremental, HOSE-code based, and neural networks. The results of the calculations are presented in Figure 2.
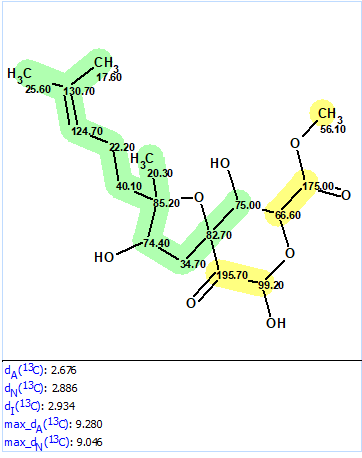
Figure 2. Originally proposed structure of asperspiropene A (1) by He et al [1] for which 13C chemical shift prediction was carried out using HOSE code-based method, neural networks, and incremental approach. Average deviations of 13C chemical shifts determined by these methods are denoted as dA, dN and dI correspondingly. Each atom is colored to mark a difference between its experimental and calculated 13C chemical shifts. The green color represents a difference between 0 to 3 ppm, yellow was >3 to 15 ppm.
Figure 2 shows that the average deviations calculated by all three methods are in the range of values which are typical for correct structures, so nothing would indicate at this stage that the structure is wrong. At the same time two definite parts of the structure are highlighted in green and yellow. In this case we can suppose that the green fragment is determined correctly while the yellow part needs to be verified. For this, the spectroscopic data obtained in [1] and presented in Table 1 were entered into ACD/SE.
Table 1. NMR spectroscopic data of asperspiropene A.
| C/X Label | δC | δC calc (HOSE) | XHn | δH | H to C HMBC |
| C 2 | 85.2 | 84.19 | C | ||
| C 3 | 74.4 | 77.2 | CH | 3.95 | |
| C 4 | 34.7 | 37.16 | CH2 | 2.16 | C 10, C 5, C 6 |
| C 4 | 34.7 | 37.16 | CH2 | 2.36 | |
| C 5 | 82.7 | 84.06 | C | ||
| C 6 | 195.7 | 204.98 | C | ||
| C 7 | 99.2 | 92.35 | CH | 5.24 | C 5 |
| C 8 | 175 | 170.85 | C | ||
| C 9 | 66.6 | 73.33 | CH | 4.51 | |
| C 10 | 75 | 77.45 | CH | 3.7 | |
| C 11 | 40.1 | 38.81 | CH2 | 1.39 | |
| C 12 | 22.2 | 22.98 | CH2 | 1.93 | |
| C 13 | 124.7 | 125.64 | CH | 5.07 | |
| C 14 | 130.7 | 131.42 | C | ||
| C 15 | 25.6 | 25.7 | CH3 | 1.62 | |
| C 16 | 17.6 | 17.65 | CH3 | 1.55 | |
| C 17 | 20.3 | 21.46 | CH3 | 0.91 | |
| C 18 | 56.1 | 52.74 | CH3 | 3.69 | |
| O 1 | OH | 5.35 |
Then a fragment was defined using the respective tool in ACD/SE (Figure 3) consisting of all the “green” atoms from Figure 2. This fragment was used to generate a User Molecular Connectivity Diagram (User MCD), with the option provided in ACD/SE (Figure 3). The resulting User MCD is shown in Figure 4.
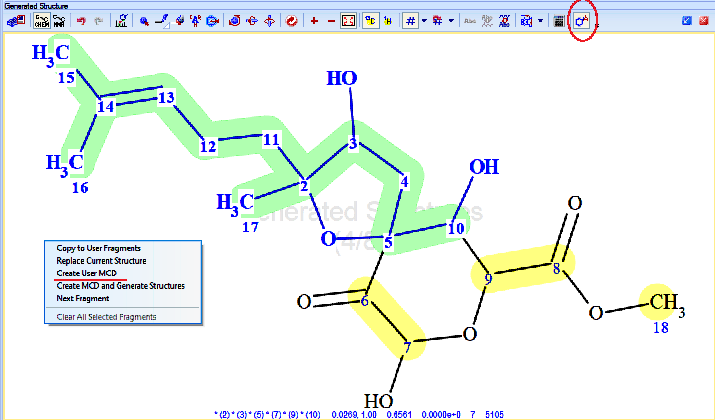
Figure 3. Fragment selection tool (red circle) and menu option for creating a User MCD based on a manually defined fragment. The selected fragment consists of the bold blue atoms and bonds, highlighted in green.
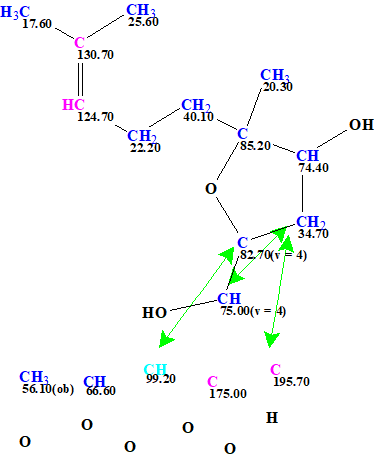
Figure 4. The User MCD of asperspiropene A. The MCD contains the fragment of the asperspiropene A structure colored with green color in Figures 2 and 3. Hybridizations of carbon atoms are marked by corresponding colors: sp2 – violet, sp3 – blue. Labels “ob” and “fb” are set by the program to carbon atoms for which neighboring with heteroatom is either obligatory (ob) or forbidden (fb).
Structure generation was run with the following output result: k=10→(filter)→8(duplicate removal)→8, tg=1 s. 13C chemical shift prediction was performed using the three mentioned approaches and the output file was ranked in increasing order of average deviations dA. The results are shown in Figure 5.
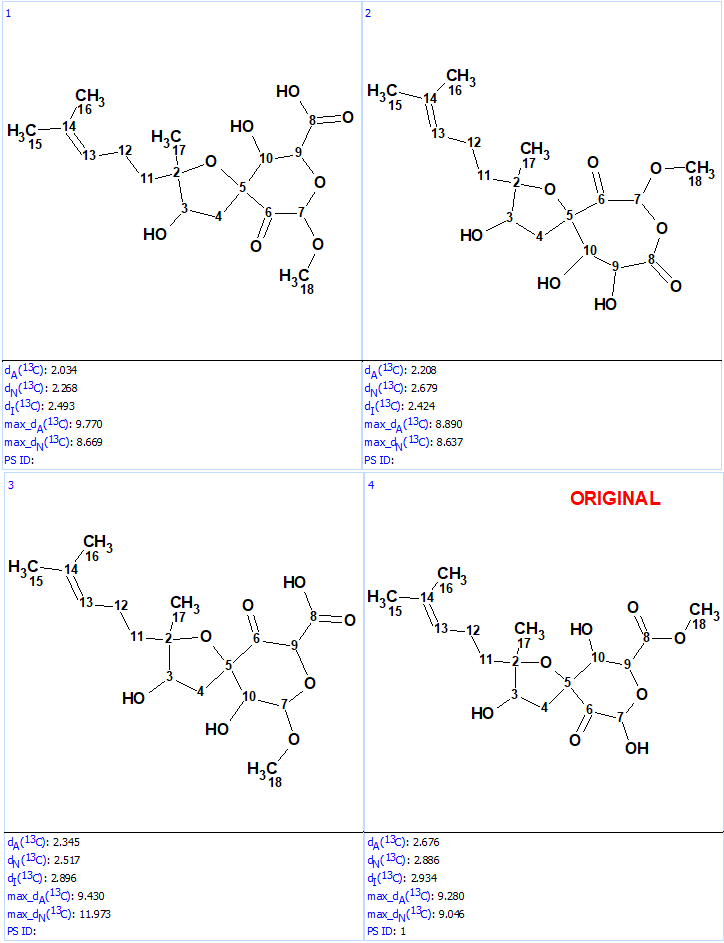
We see that structures #1- #6 all have acceptable average deviations, while the originally proposed structure is placed at the 4th position by the ranking procedure. To evaluate the solution obtained, the IR spectrum of the compound as was reported in [1] was examined. All the generated structures contain an unconjugated ketone group as well as either an acid or ester moiety. Therefore, we can expect that strong absorption will be observed in the region of 1700-1740 cm-1 of the IR spectrum. However, as seen in Figure 6 there are no bands in this area, which is clear evidence of the fact that all the structures of the output file are incorrect.
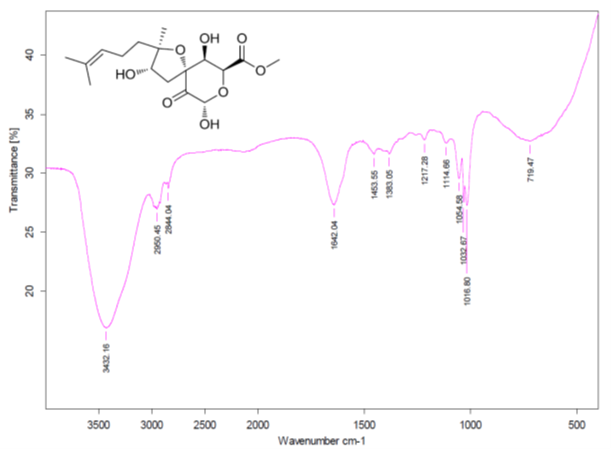
Figure 6. Infrared spectrum of compound 1 as presented in the Supplementary Information of [1].
This clearly indicates that a revision of the data is required, and this is what the authors of [2] did. They carefully examined the HR-ESI-MS of the isolated compound and recognized that the molecular ion was incorrectly identified in the original article [1]. Therefore the correct molecular formula of asperspiropene A is C17H26O6, containing two atoms of oxygen less than the originally reported MF.
After this the procedure in ACD/SE described above wasrepeated but with the corrected MF this time. The resulting User MCD is shown in Figure 7.
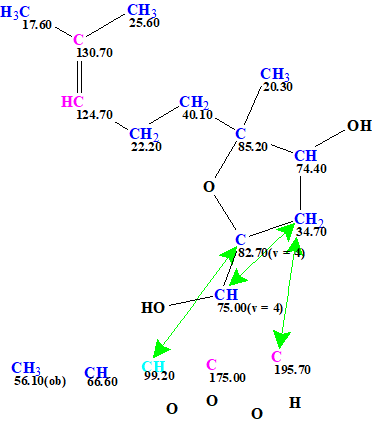
Figure 7. User MCD created from the revised molecular formula C17H26O6. Note that still the originally reported incorrect structure was used as a source to define the fragment used, this is not a problem at all for ACD/SE.
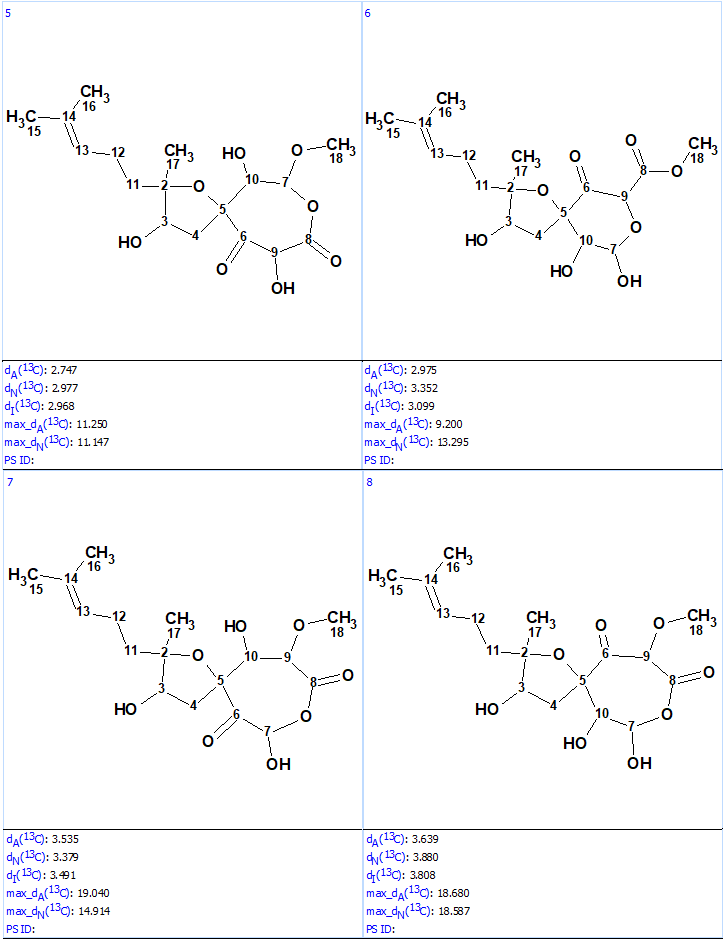
Structure generation from the new User MCD was completed with the following results: k = 52→(filtering)→8→(duplicate removal)→8, tg = 1 s. The ranked file with the structures is presented in Figure 8.
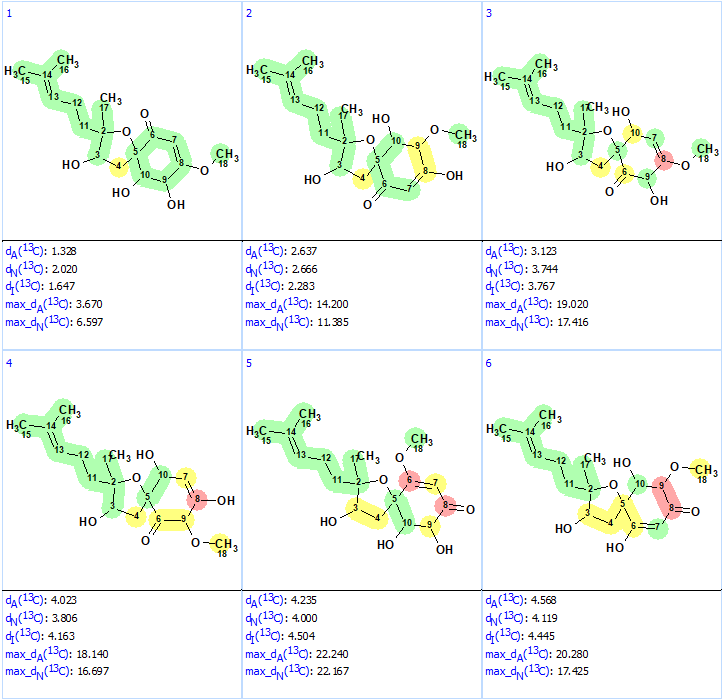
Figure 8. Ranked structures file generated for the revised molecular formula. The red color represents a difference >15 ppm between experimental and calculated chemical shifts.
We see that now the revised structure was placed at the first position by the ranking procedure. A single yellow carbon atom has deviation between experimental and calculated values equal only to 3.7 ppm, which additionally confirms the validity of the best structure. The presence of a conjugated ketone in the structure is in agreement with the IR band observed at 1640 cm-1. Figure 9 shows the DP4 probabilities calculated for structurers #1 and #2. We see that the mean deviations for the first structure in Figure 9 are much lower than the ones for the originally reported incorrect structure as well as all the other structures generated with the incorrect MF (Figure 5).
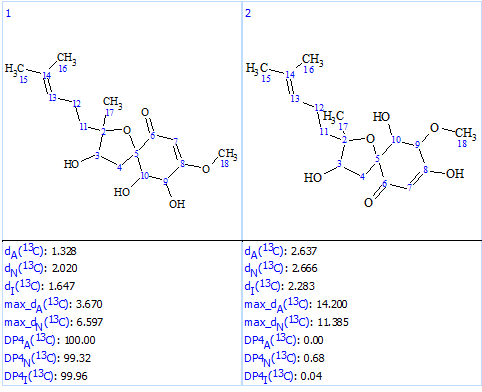
Figure 9. DP4 probabilities calculated for structures #1 and #2.
We see that the use of ACD/SE allows us to convincingly reject the originally reported structure and determine the revised, correct one. This required the careful analysis of the IR and HR-MS data.
This example illustrates that a structure can be found to be wrong regardless of the NMR data. We had structures generated that were in good agreement with the observed experimental data but disagreed with the IR data. This alone should had raised some suspicions to the researchers and directed them to look at the situation more closely, and maybe re-examining that HR-MS spectrum. Dismissing experimental data that is not agreeable to what we want or expect is never good. Unfortunately, NMR spectroscopists frequently ignore structural information which can be extracted easily from vibrational spectra (IR and Raman) and focus only on NMR, which can lead to problems.
References
- He, Y.; Zheng, M.; Li, Q.; Hu, Z.; Zhu, H.; Liu, J.; Wang, J.; Xue, Y.; Li, H.; Zhang, Y. (2017). Asperspiropene A, a novel fungal metabolite as an inhibitor of cancer-associated mutant isocitrate dehydrogenase. Org. Chem. Front., 4(6), 1137−1144.
- V. A Cao, B.-K. Choi, H.-S. Lee, C.-S. Heo, H. J. Shin. (2021). Reisolation and Structure Revision of Asperspiropene A. J. Nat. Prod., 84, 1843−1847.
About the Author
