October 1, 2021
by Mikhail Elyashberg, Leading Researcher, ACD/Labs
Lyaline. Structure revision
The original sample of lyaline was isolated from the roots of a Madagascan rubiaceous plant, Pauridiantha paucinervis in 1974. It was then reported to have a unique harman-1,4-dihydropyridine scaffold (1). That structure was determined only from EIMS and incomplete 1H NMR data, as recording 13C NMR spectra was not routinely available at that time.
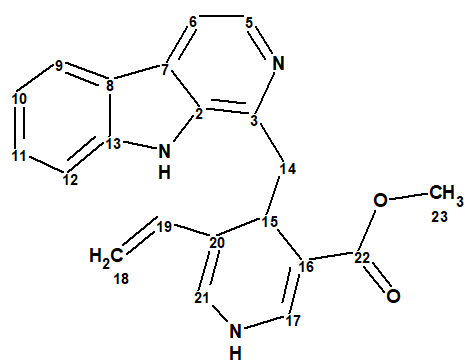
1
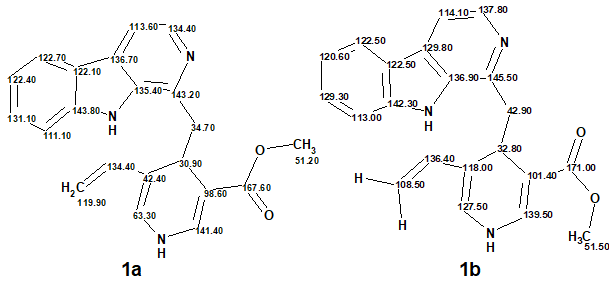
Thirty years later a total synthesis of the claimed structure of lyaline revealed critical inconsistencies with the spectroscopic data initially reported (Figure 1):
1
Figure 1. Comparison of 13C chemical shift assignments corresponding to the original structure (1a) with those measured for the synthesized structure (1b).
The synthesis of the originally proposed structure also revealed that this dihydropyridine compound is prone to facile fragmentation into harmane and pyridine, rendering it unlikely to withstand the harsh alkaloidal fractionation procedure used. Re-isolation was not an option for revising the lyaline structure as the spectroscopic data related to the original sample were not complete enough to reliably ascertain its identity with new isolates. Finding the original sample of lyaline enabled Jagora at al[1] to perform new spectroscopic analysis of this compound using present state-of-the-art techniques.
The unique peak observed in the LC-MS revealed a signal at m/z 346.1551, consistent with the expected elemental composition of C21H19N3O2. The stability of the sample, however, was inconsistent with the fragility of the harman-1,4-dihydropyridine synthesized earlier.
As a result of the compound reinvestigation using 1D and 2D NMR (HSQC, COSY, TOCSY, 1H-13C HMBC and 1H-15N HMBC) data the correct structure of lyaline was determined:
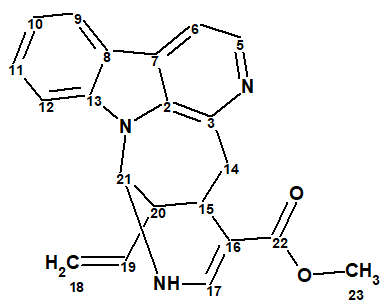
2
We utilized the available from the article key 2D NMR correlations (Table 1) to show how the structure revision could be carried out with the assistance of ACD/Structure Elucidator (ACD/SE).
Table 1. NMR spectroscopic data obtained in work [1].
| C/N Label | δC | δC calc | δN | XHn | δH | M | COSY | H to C HMBC | HN HMBC |
| C 2 | 135.4 | 135.09 | C | ||||||
| C 3 | 143.2 | 142.26 | C | ||||||
| C 5 | 134.4 | 137.3 | CH | 8.31 | d | 7.91 | C 7, C 3 | ||
| C 6 | 113.6 | 113.51 | CH | 7.91 | d | 8.31 | C 8, C 2 | ||
| C 7 | 136.7 | 130.63 | C | ||||||
| C 8 | 122.1 | 120.81 | C | ||||||
| C 9 | 122.7 | 120.34 | CH | 8.15 | d | 7.4 | C 7, C 13 | ||
| C 10 | 122.4 | 119.66 | CH | 7.4 | t | 7.70, 8.15 | |||
| C 11 | 131.1 | 126.39 | CH | 7.7 | t | ||||
| C 12 | 111.1 | 110.43 | CH | 7.7 | d | 7.4 | C 13 | ||
| C 13 | 143.8 | 138.83 | C | ||||||
| C 14 | 34.7 | 36.4 | CH2 | 3.57 | u | 3.52 | C 16, C 3 | ||
| C 14 | 34.7 | 36.4 | CH2 | 4.02 | u | ||||
| C 15 | 30.9 | 36.92 | CH | 3.52 | u | 3.39, 3.57 | C 16, C 3, C 22 | ||
| C 16 | 98.6 | 96.04 | C | ||||||
| C 17 | 141.4 | 140.48 | CH | 7.16 | u | 6.21 | C 15, C 16, C 22 | N 3 | |
| C 18 | 119.9 | 119.28 | CH2 | 5.34 | u | ||||
| C 18 | 119.9 | 119.28 | CH2 | 5.27 | u | 6 | |||
| C 19 | 134.4 | 133.13 | CH | 6 | u | 3.39, 5.27 | |||
| C 20 | 42.4 | 45.17 | CH | 3.39 | u | 3.52, 6.00, 6.13 | N 1 | ||
| C 21 | 63.3 | 62.28 | CH | 6.13 | u | 3.39, 6.21 | C 2, C 13 | N 1 | |
| C 22 | 167.6 | 168.38 | C | ||||||
| C 23 | 51.2 | 50.51 | CH3 | 3.6 | u | C 22 | |||
| N 1 | 136 | N | |||||||
| N 2 | 227 | N | |||||||
| N 3 | 96 | NH | 6.21 | u | 6.13, 7.16 | C 17 |
First, spectroscopic data were entered into ACD/SE and structure 1a (Figure 1) was placed into the Proposed Structure, PS, container of the program. 13C chemical shift prediction was performed for structure 1a by means of the three approaches incorporated into the ACD/SE (HOSE code-based, neural networks and incremental method). Figure 2 shows structure 1a and the average deviations between experimental and predicted chemical shifts for the three prediction methods.
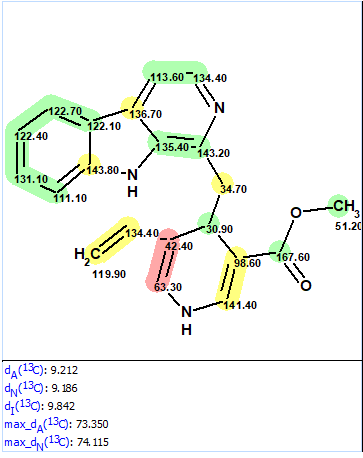
Figure 2. Original structure 1a.
Figure 2 also shows that the upper part of the structure is confirmed by the chemical shift prediction while the lower one displays extremely large deviations for carbon atoms C 42.4 and C 63.3. It is evident that the structure is wrong.
To produce all possible candidate structures, the following approach available in ACD/SE was utilized: The upper fragment of structure 1a was selected in the structure and an MCD containing this fragment was created. The mentioned procedure is illustrated by Figure 3, and the automatically created MCD is presented in Figure 4.
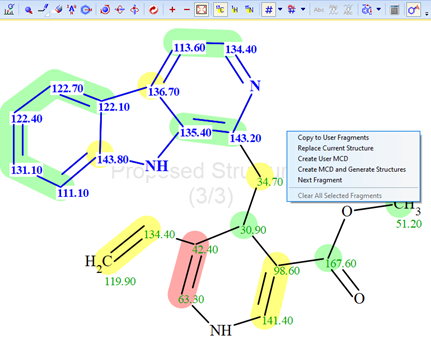
Figure 3. “Cutting”, selecting the upper fragment of the molecule followed by selection of the command “Create User MCD”.
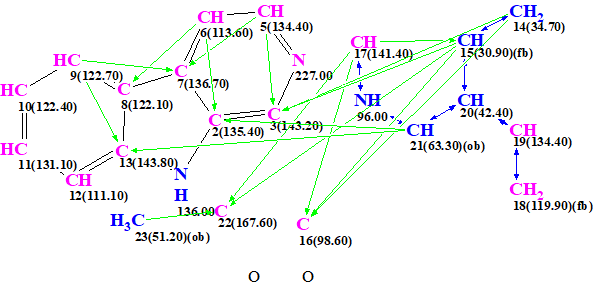
Figure 4. Automatically created MCD containing the upper fragment of structure 1a.
As a result of structure generation from the MCD a single structure was produced in 0.4 s (Figure 5).
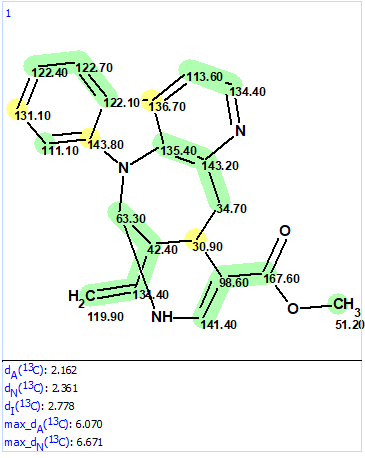
Figure 5. Resulting structure from MCD using user fragment
We see that the structure coincides with structure 2 and both values of the average deviations and colors of carbon atoms illustrate the structure validity.
This was also confirmed by structure generation from an MCD (Figure 6) created on the basis of the spectroscopic data collected in Table 1 alone, without a pre-defined fragment.
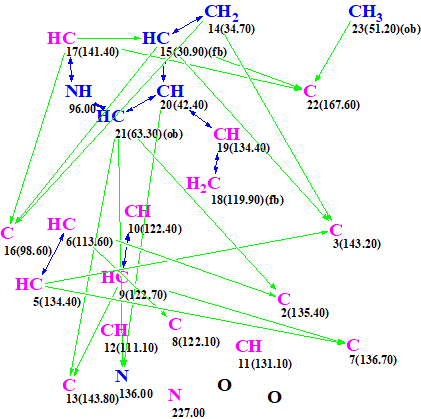
Figure 6. Molecular connectivity diagram created from 1D and 2D NMR data presented in Table 1.
The structure generation was completed with the following result: k=12 → (structure filtering) → 3, tg = 0.5 s. The output file ranked in increasing order of average deviations is shown in Figure 7.
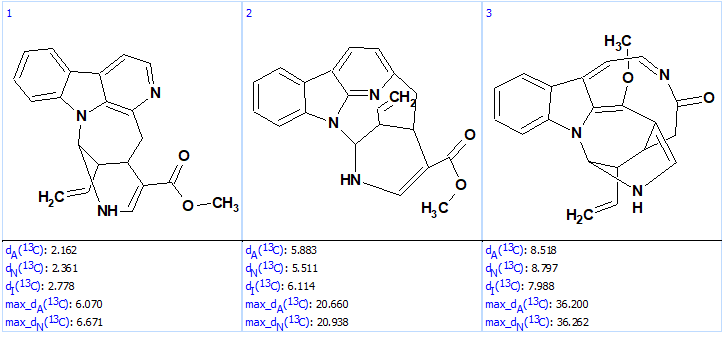
Figure 7. Ranked output file.
Figure 7 shows that the validity of the revised structure of lyaline is reliably confirmed by “ab initio“, “de novo” solving, that is by structure generation from the molecular formula and NMR spectroscopic data alone without biasing from user fragments. Interestingly, the potentially time-consuming acquisition of the 1H-15N HMBC spectrum is not required if ACD/SE is used. When this spectrum is excluded from the NMR data set the following result are obtained:
k=162 → (structure filtering) → 78 → (duplicate removal) → 38, tg =5 s, and the revised structure was again the top ranked one (Figure 8).
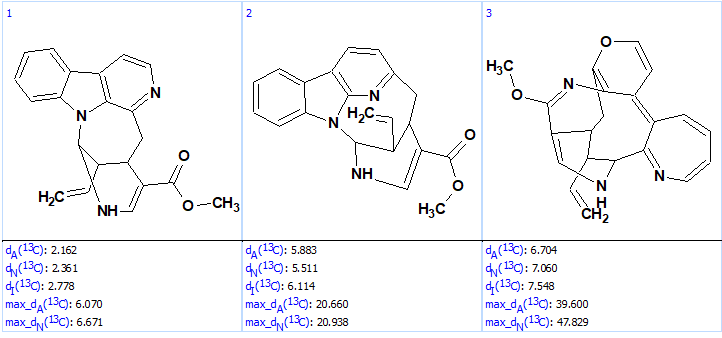
Figure 8. Top ranked structures of the output file.
In conclusion, the structure of lyaline was quickly and convincingly re-elucidated with the assistance of a CASE program, ACD/SE, which is not only a powerful tool for structure elucidation but also for structure verification and revision. You can find similar examples of ACD/SE use for structure revision in previous publications [2-3] and in a more recent one [4].
References
- Jagora, A., Gallard, J.-F., Beniddir, M. A., Le Pogam, P. (2021). A Reappraisal of the Structure of Lyaline as the First Naturally Occurring Nacycline Monoterpene Indole Alkaloid. J. Nat. Prod., 84(9), 2617–2622. https://doi.org/10.1021/acs.jnatprod.1c00572
- Elyashberg, M., Williams, A., Blinov, K. (2010). Structural revisions of natural products by Computer Assisted Structure Elucidation (CASE) Systems. Nat. Prod. Rep., 27(9), 1296–1328. https://doi.org/10.1039/C002332A
- Elyashberg, M., Blinov, K., Molodtsov, S., Williams, A.J. (2013). Structure Revision of Asperjinone Using Computer-Assisted Structure Elucidation Methods. J. Nat. Prod., 76, 113–116. https://pubs.acs.org/doi/10.1021/np300218g
- Lv, T.-M., Chen, D.-L., Liang, J.-J., Bai, M., Lin, B., Huang, X.-X., Ma, G.-X., Song, S.-J. (2021). Structural Revisions of Two Highly Aromatic Naphthoquinone-Derived Dimers Based on NMR Analysis, Computer-Assisted Structure Elucidation Methods, and Computations. Organic Letters, 23(18), 7231–7235. https://doi.org/10.1021/acs.orglett.1c02626
About the Author
